

Aproximadamente un 20-40% de los casos de hipercolesterolemia familiar diagnosticada no muestran mutación causal en los genes candidatos, por lo que algunos de estos casos pueden tener un origen poligénico. Se han identificado diferentes variantes genéticas de un solo nucleótido que ayudan a diferenciar las hipercolesterolemias poligénicas de las monogénicas. El objetivo es estudiar la contribución de dichas variantes a la concentración de colesterol unido a lipoproteínas de baja densidad (cLDL) en probandos con hipercolesterolemia genética sin mutación en genes candidatos (hipercolesterolemia genética sin hipercolesterolemia familiar) y establecer el valor de una puntuación genética basada en las frecuencias de dichas variantes de un solo nucleótido en el cribado en cascada de sus familiares.
MétodosSe reclutó a 49 familias con hipercolesterolemia genética sin hipercolesterolemia familiar (294 sujetos) y se calculó la puntuación genética derivada de las variantes de un solo nucleótido de los genes SORT1, APOB, ABCG8, APOE y LDLR más la concentración plasmática de lipoproteína(a).
ResultadosLos alelos de riesgo en SORT1, ABCG8, APOE y LDLR presentaron mayor frecuencia en los consanguíneos que en el proyecto 1.000 Genomas, con diferencia estadísticamente significativa. La contribución de la puntuación genética a la concentración plasmática de cLDL fue significativamente mayor en los sujetos afectados de hipercolesterolemia que en los de control (p=0,048). El porcentaje de la variación de cLDL explicado por la puntuación fue del 3,1%, que aumentó al 6,9% seleccionando a las familias con puntuación genética más alta en el probando.
ConclusionesLas familias con hipercolesterolemia genética sin hipercolesterolemia familiar concentran los alelos de riesgo de cLDL alto. Su contribución varía mucho entre las familias, lo que indica la complejidad y la heterogeneidad de estas formas de hipercolesterolemia. La puntuación genética explica un pequeño porcentaje del cLDL, lo que limita su uso diagnóstico.
Palabras clave
La hipercolesterolemia familiar (HF) es un trastorno genético caracterizado por unas concentraciones de colesterol total en plasma muy altas, a causa de un aumento del colesterol unido a lipoproteínas de baja densidad (cLDL), con un riesgo alto de enfermedad coronaria prematura1. Tradicionalmente, la HF se ha descrito como una enfermedad monogénica de transmisión autosómica codominante con una prevalencia estimada en alrededor de 1:500 en la población general1. Estudios recientes han revelado que la HF clínicamente definida probablemente sea más frecuente de lo que se había descrito con anterioridad, con una prevalencia de 1:217 en el estudio Copenhagen General Population, en el que se analizó a población general2. Esta tasa de prevalencia llega a ser de 1:70 en algunas poblaciones con efecto de gen fundador, como la de los afrikáneres de Sudáfrica3. La HF tiene origen en mutaciones en los genes LDLR, que es el que codifica el receptor de las LDL; APOB, que codifica la apolipoproteína B4, y PCSK9, que codifica la enzima proproteína convertasa subtilisina/kexina tipo 95. Se han identificado 2 nuevos loci a los que se atribuye la HF: La mutación p.(Leu167del) en el gen APOE6 y varias mutaciones en el miembro de la familia del adaptador de transductor de señal STAP17. Sin embargo, no se observa ninguna mutación causal en los genes candidatos en aproximadamente un 20-40% de los casos de HF clínicamente establecidos8. Las posibles explicaciones de estos datos son la existencia de otros genes no descubiertos, a pesar de los amplios estudios negativos mediante análisis de secuenciación del exoma9, la falta de especificidad de los criterios diagnósticos clínicos actuales para el diagnóstico de la HF en la identificación de un trastorno monogénico y el hecho de que el fenotipo lipídico y la forma de presentación familiar dentro de la familia de algunas hipercolesterolemias poligénicas se solapan por completo con la HF definida genéticamente. Esto último parece ser lo que sucede en muchos pacientes con HF clínicamente definida, tal como han demostrado con pulcritud Talmud et al.10. De manera en todo coherente con el trasfondo poligénico de algunas HF clínicamente definidas, nuestro equipo ha estudiado recientemente un grupo de familias con un diagnóstico clínico de HF, pero sin una mutación causal en los genes candidatos; en esas familias, los resultados de la segregación familiar y la heredabilidad del colesterol fueron compatibles con una enfermedad poligénica en vez de monogénica. Por consiguiente, el término hipercolesterolemia genética sin HF (HGSHF) parece una designación más apropiada para este tipo de hipercolesterolemia11. La caracterización del componente genético, monogénico o poligénico, de una hipercolesterolemia específica puede tener consecuencias clínicas, entre ellas el uso de la detección genética en cascada, el asesoramiento genético o la evaluación del riesgo de enfermedad coronaria, así como en cuestiones administrativas relacionadas con la prescripción o el reembolso de determinados fármacos que están indicados especialmente para la HF monogénica12.
Se ha demostrado que una puntuación del riesgo genético del colesterol para el diagnóstico de la hipercolesterolemia poligénica tiene utilidad para diferenciar los casos de hipercolesterolemia poligénica de los monogénicos, y su uso se ha validado en diferentes cohortes de Europa, Canadá, Israel y Corea10-13. Sin embargo, esta puntuación genética no se ha estudiado anteriormente en familias con sospecha de hipercolesterolemia monogénica. En muchas personas con HGSHF hay unos antecedentes familiares claros de hipercolesterolemia y, por consiguiente, los estudios familiares serían muy útiles para confirmar la contribución a la acumulación de alelos que elevan el cLDL frecuentes y de efecto pequeño como causa de la hipercolesterolemia en determinadas familias, así como establecer si esta puntuación puede ser útil para identificar a los familiares afectados en un examen de detección en cascada. Así pues, se calculó la puntuación génica del colesterol basada en 6 frecuentes variantes de un solo nucleótido (SNV) que aumentan el cLDL, que se encuentran en 5 genes diferentes, así como las concentraciones plasmáticas de lipoproteína(a), un tipo de lipoproteína determinado genéticamente que contribuye a aumentar la concentración de colesterol, en una muestra de 49 familias con HGSHF, es decir, con un diagnóstico clínico de HF, pero sin una mutación causal en los genes candidatos de la HF.
MÉTODOSParticipantesEl protocolo se ha publicado ya con anterioridad11. De manera resumida, se invitó a participar en este estudio de familias a participantes con HGSHF consecutivos. Los criterios de inclusión para los probandos fueron los siguientes: edad> 18 años; colesterol total y cLDL por encima del percentil 95 y triglicéridos por debajo del percentil 90 según la distribución por edad y sexo de la población española13, al menos 1 familiar de primer grado con cLDL superior al percentil 90 y> 6 puntos según los criterios de la Dutch Lipid Clinic Network12, 3 familiares de primer grado vivos y ausencia de mutaciones patógenas de la HF en los genes LDLR, APOB y PCSK9 estudiados con la plataforma Lipochip8, una plataforma de diagnóstico genético, una micromatriz para la detección en estos 3 genes de mutaciones españolas frecuentes, incluida la variación en el número de copias de LDLR y reordenaciones grandes, seguido de un análisis de secuenciación de las regiones de codificación de LDLR y el exón 26 de APOB cuando los resultados eran negativos. Los criterios de exclusión de los probandos fueron las hipercolesterolemias secundarias y la presencia del genotipo APOE ¿2/¿2 o la mutación p.(Leu167del) en APOE. A partir de cada probando seleccionado, se intentó reclutar el máximo número de familiares posible, incluidos padres, hermanos, cónyuges, hijos y sobrinos. Antes de que se aplicara algún método de la investigación, todos los participantes firmaron un documento de consentimiento informado aprobado por el comité local de ética de investigación (Comité Ético de Investigación de Aragón). La hipercolesterolemia en los familiares se definió por la presencia de valores de cLDL superiores al percentil 90 correspondiente por edad y sexo14.
Evaluaciones clínicas y analíticasSe registraron los antecedentes personales y familiares de enfermedad cardiovascular, uso de medicación y factores de riesgo cardiovascular de los probandos y los familiares. Se obtuvieron de todos los participantes muestras de suero y plasma con ácido etilendiaminotetraacético, tras un mínimo de 10 h en ayunas y un periodo de 6 semanas sin uso de medicación hipolipemiante. Se determinaron las concentraciones de colesterol total y triglicéridos con métodos enzimáticos estándares. El colesterol unido a lipoproteínas de alta densidad se determinó directamente con una reacción enzimática utilizando colesterol oxidasa (UniCel DxC 800, Beckman Coulter Inc.; Brea, California, Estados Unidos). Las determinaciones de lipoproteína(a), apolipoproteína A1, apolipoproteína B y proteína C reactiva se realizaron con la nefelometría cinética IMMAGE (Beckman Coulter Inc.). El cLDL se calculó con la fórmula de Friedewald.
Análisis genéticoSe aisló el ADN genómico de muestras de sangre total mediante métodos estándares. Se genotipificaron las SNV de los genes SORT1, APOB, ABCG8 y LDLR con sondas TaqMan por métodos estándares. Se determinó el genotipo APOE mediante secuenciación de ADN del exón 4, según lo descrito anteriormente15.
Análisis estadísticoPara los análisis se utilizó el programa SPSS versión 20.0 (Chicago, Illinois, Estados Unidos). El valor nominal de significación fue p<0,05. Se analizó la distribución normal de las variables mediante la prueba de Kolmogorov-Smirnov. Las variables cuantitativas con distribución normal se expresaron en forma de media±desviación estándar y se analizaron con la prueba de la t de Student. Las variables con distribución asimétrica se expresaron en forma de mediana [intervalo intercuartílico] y se analizaron con la prueba de la U de Mann-Whitney. Las variables cualitativas se expresaron en forma de porcentaje y se analizaron con la prueba de la χ2. Para comparar la frecuencia alélica de las variantes genéticas, se utilizamos la prueba de la χ2 para comparar los alelos de tipo natural (wild) con los mutantes. La asociación del cLDL con las SNV y la puntuación genética se analizó mediante una regresión lineal que incluía el índice de masa corporal, el sexo, la edad y el perímetro de cintura como factores de confusión.
El tamaño muestral se estableció considerando una media de puntuación génica del cLDL en la HF de 0,708±0,19 y la media de puntuación génica de los controles de 0,632±0,22)13. Se estableció un nivel de confianza (1 – α) del 95% (Zα unilateral=1,960) con una potencia estadística (1 – β) del 90% (Zβ unilateral=1,282), con lo que se obtuvo un tamaño muestral de 126 participantes tras introducir un ajuste para un 15% de pérdidas previstas.
Puntuación génica del colesterolSe calculó para cada individuo la puntuación génica del colesterol mediante la suma ponderada de los alelos de riesgo de SORT1, APOB, ABCG8, LDLR y APOE y de la concentración de lipoproteína(a).
Anteriormente se ha demostrado que estas SNV presentan una asociación intensa con la hipercolesterolemia poligénica. La ponderación utilizada para cada alelo fue el coeficiente beta (de riesgo) por alelo descrito por el Global Lipids Genetics Consortium (tabla 1)10. El cálculo del valor del colesterol transportado en la lipoproteína(a) se hizo según lo recomendado por Dahlen16,17: concentración de lipoproteína(a)=0,3×lipoproteína(a) en mg/dl, y se sumó al resultado de la puntuación genética.
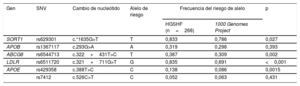
Ponderación del Global Lipids Genetics Consortium para las 6 variantes de un solo nucleótido utilizadas para el cálculo de la puntuación génica del colesterol*
| Gen | SNV | Cambio de nucleótido | Alelo de riesgo | Ponderación de GLGC para el cálculo de la puntuación (mg/dl) |
|---|---|---|---|---|
| SORT1 | rs629301 | c.*1635T>G | T | +5,850 |
| APOB | rs1367117 | c.293G>A | A | +3,867 |
| ABCG8 | rs6544713 | c.322+431T>C | T | +2,769 |
| LDLR | rs6511720 | c.321+711G>T | G | +7,020 |
| APOE | rs429358 | c.388T>C | C | |
| rs7412 | c.526C>T | T | ||
| ¿2/¿3 | –15,6 | |||
| ¿2/¿4 | –7,8 | |||
| ¿3/¿3 | 0 | |||
| ¿3/¿4 | +3,9 | |||
| ¿4/¿4 | +7,8 |
A: adenina; C: citosina; G: guanina; GLGC: Global Lipids Genetics Consortium; SNV: variante de un solo nucleótido; T: timina.
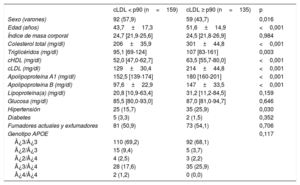
Durante el periodo de estudio, se examinó en total a 1.648 pacientes no emparentados con un diagnóstico clínico de hipercolesterolemia genética primaria, y hubo 243 probandos que cumplían los criterios de inclusión. Se invitó consecutivamente a los que iban cumpliendo los criterios de inclusión a participar hasta que se alcanzó el número previsto de 50 familias. Después de la caracterización inicial de los probandos, se excluyó a 1 familia a causa de una asignación compleja de la paternidad. Se incluyó en total a 294 participantes de las 49 familias estudiadas: 268 familiares directos (91,2%) y 26 cónyuges (8,8%). Los familiares con hipercolesterolemia eran de más edad, incluían un mayor porcentaje de mujeres y tenían valores de colesterol total, cLDL y colesterol unido a lipoproteínas de alta densidad más altos que los familiares sin hipercolesterolemia. Las características clínicas y antropométricas de estos participantes en función de la presencia o ausencia de hipercolesterolemia se indican en la tabla 2.
Características bioquímicas y clínicas de los familiares con cLDL <percentil 90 y los participantes con cLDL ≥ percentil 90*
| cLDL < p90 (n=159) | cLDL ≥ p90 (n=135) | p | |
|---|---|---|---|
| Sexo (varones) | 92 (57,9) | 59 (43,7) | 0,016 |
| Edad (años) | 43,7±17,3 | 51,6±14,9 | <0,001 |
| Índice de masa corporal | 24,7 [21,9-25,6] | 24,5 [21,8-26,9] | 0,984 |
| Colesterol total (mg/dl) | 206±35,9 | 301±44,8 | <0,001 |
| Triglicéridos (mg/dl) | 95,1 [69-124] | 107 [83-161] | 0,003 |
| cHDL (mg/dl) | 52,0 [47,0-62,7] | 63,5 [55,7-80,0] | <0,001 |
| cLDL (mg/dl) | 129±30,4 | 214±44,8 | <0,001 |
| Apolipoproteína A1 (mg/dl) | 152,5 [139-174] | 180 [160-201] | <0,001 |
| Apolipoproteína B (mg/dl) | 97,6±22,9 | 147±33,5 | <0,001 |
| Lipoproteína(a) (mg/dl) | 20,8 [10,9-63,4] | 31,2 [11,2-84,5] | 0,159 |
| Glucosa (mg/dl) | 85,5 [80,0-93,0] | 87,0 [81,0-94,7] | 0,646 |
| Hipertensión | 25 (15,7) | 35 (25,9) | 0,030 |
| Diabetes | 5 (3,3) | 2 (1,5) | 0,352 |
| Fumadores actuales y exfumadores | 81 (50,9) | 73 (54,1) | 0,706 |
| Genotipo APOE | 0,117 | ||
| ¿3/¿3 | 110 (69,2) | 92 (68,1) | |
| ¿2/¿3 | 15 (9,4) | 5 (3,7) | |
| ¿2/¿4 | 4 (2,5) | 3 (2,2) | |
| ¿3/¿4 | 28 (17,6) | 35 (25,9) | |
| ¿4/¿4 | 2 (1,2) | 0 (0,0) |
cHDL: colesterol unido a lipoproteínas de alta densidad; cLDL: colesterol unido a lipoproteínas de baja densidad.
Las variables cuantitativas se expresan en forma de media±desviación estándar, excepto por las variables que no muestran una distribución normal, que se expresan en forma de mediana [intervalo intercuartílico]. Las variables cualitativas se expresan en forma de n (%). El valor de p se calculó con la prueba de la t de Student o con la prueba de la U de Mann-Whitney y con pruebas de la χ2, según procediera.
Todos los alelos de riesgo mostraron mayor frecuencia en las familias con HGSHF que en el 1000 Genomes Project18, si bien las diferencias fueron estadísticamente significativas solo en 4 de ellas: c.*1635G>T en el SORT1, c.322+431T>C en ABCG8, c.327+711 G>T en LDLR y c.388T>C en APOE (tabla 3). Las frecuencias de los alelos de riesgo no mostraron diferencias significativas entre los participantes con y sin hipercolesterolemia dentro de las familias. Hubo mayor frecuencia de todas las SNV en los participantes afectados, aunque no se alcanzó una diferencia estadísticamente significativa entre los participantes afectados y los no afectados en las familias con HGSHF. Las frecuencias de alelos de todas las SNV siguieron el equilibrio de Hardy-Weinberg. Sin embargo, la puntuación génica del colesterol fue significativamente mayor (p=0,048) en los participantes con cLDL> percentil 90 en comparación con los de cLDL <percentil 90 (). Al dividir a los participantes en cuartiles según la puntuación génica del colesterol (tabla 4), hubo un aumento significativo del cLDL con los cuartiles más altos de la puntuación génica del colesterol (p=0,007). Cada aumento de aproximadamente 1 punto se acompañaba de un aumento del cLDL de 1 mg/dl, y cada cuartil difería en aproximadamente 10 puntos.
Frecuencia alélica de las variantes genéticas en familiares directos de personas con hipercolesterolemia genética sin hipercolesterolemia familiar y en el 1000 Genomes Project
| Gen | SNV | Cambio de nucleótido | Alelo de riesgo | Frecuencia del riesgo de alelo | p | |
|---|---|---|---|---|---|---|
| HGSHF (n=268) | 1000 Genomes Project | |||||
| SORT1 | rs629301 | c.*1635G>T | T | 0,833 | 0,786 | 0,027 |
| APOB | rs1367117 | c.293G>A | A | 0,319 | 0,298 | 0,393 |
| ABCG8 | rs6544713 | c.322+431T>C | T | 0,387 | 0,309 | 0,002 |
| LDLR | rs6511720 | c.321+711G>T | G | 0,835 | 0,691 | <0,001 |
| APOE | rs429358 | c.388T>C | C | 0,138 | 0,086 | 0,0015 |
| rs7412 | c.526C>T | C | 0,052 | 0,063 | 0,431 | |
A: adenina; C: citosina; G: guanina; HGSHF: hipercolesterolemia genética sin hipercolesterolemia familiar; SNV: variante de un solo nucleótido; T: timina.
Los valores de p se calcularon con la prueba de la χ2, comparando las frecuencias alélicas de mutantes con las del tipo natural (wild).
Concentraciones de colesterol unido a lipoproteínas de baja densidad según los cuartiles de la puntuación génica del colesterol en familiares directos de pacientes con hipercolesterolemia genética sin hipercolesterolemia familiar
| Cuartil de la puntuación génica de colesterol* | cLDL explicado por la puntuación génica del colesterol (mg/dl) | cLDL medido (mg/dl) | p para la tendencia |
|---|---|---|---|
| 1 | 25,2 [20,0-28,9] | 158±55,4 | 0,007 |
| 2 | 35,6 [34,2-37,2] | 169±54,1 | |
| 3 | 41,6 [40,1-44,3] | 174±55,6 | |
| 4 | 55,7 [50,9-66,2] | 185±64,5 |
cLDL: colesterol de lipoproteínas de baja densidad.
Las variables cuantitativas se expresan como media±desviación estándar, excepto las variables que no muestran distribución normal, que se expresan como mediana [intervalo intercuartílico]. La p para la tendencia se refiere a las diferencias en el cLDL entre cuartiles de puntuación.
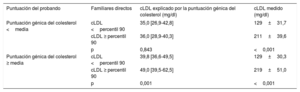
Se estudiaron las repercusiones de la puntuación génica del colesterol según la puntuación génica del colesterol existente en el probando. Se dividió a las familias en 2 grupos según el valor de la puntuación del probando: familias con puntuación génica del colesterol del probando elevada y familias con puntuación génica del colesterol del probando baja. La puntuación no mostró una asociación significativa con la hipercolesterolemia en las familias en las que la puntuación génica del colesterol del probando era baja. Sin embargo, en las familias con puntuación génica de colesterol del probando alta, la puntuación tuvo un alto valor para discriminar hipercolesterolemia en los familiares (p=0,001) (tabla 5).
Puntuación génica de colesterol y concentraciones de colesterol en familiares directos de personas con hipercolesterolemia genética sin hipercolesterolemia familiar según la puntuación génica de colesterol del probando
| Puntuación del probando | Familiares directos | cLDL explicado por la puntuación génica del colesterol (mg/dl) | cLDL medido (mg/dl) |
|---|---|---|---|
| Puntuación génica del colesterol <media | cLDL <percentil 90 | 35,0 [26,9-42,8] | 129±31,7 |
| cLDL ≥ percentil 90 | 36,0 [28,9-40,3] | 211±39,6 | |
| p | 0,843 | <0,001 | |
| Puntuación génica del colesterol ≥ media | cLDL <percentil 90 | 39,8 [36,6-49,5] | 129±30,3 |
| cLDL ≥ percentil 90 | 49,0 [39,5-62,5] | 219±51,0 | |
| p | 0,001 | <0,001 |
cLDL, colesterol de lipoproteínas de baja densidad.
El cLDL se expresa en forma de media±desviación estándar, la puntuación génica del colesterol se expresa en forma de mediana [intervalo intercuartílico]. El valor de p se calculó con la prueba de la t de Student y la prueba de la U de Mann-Whitney, según procediera.
La asociación entre el cLDL y cada SNV se analizó mediante un análisis de regresión lineal univariable. Solamente las SNV de APOB (c.293G>A) y APOE (c.526C>T) mostraron una asociación estadísticamente significativa con la concentración de cLDL al introducirlas juntas en el mismo modelo. La relación continuó siendo significativa tras introducir un ajuste respecto a los factores de confusión ().
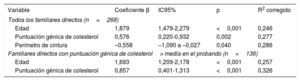
La regresión lineal mostró que la parte de la concentración de cLDL que se explicaba por la edad, la puntuación genética y el perímetro de cintura era el 28,6% tras ajustar por sexo e índice de masa corporal. El porcentaje explicado por la puntuación era de solo un 3,1%; sin embargo, aumentaba al 6,9% en el grupo de participantes con la puntuación máxima en el probando (tabla 6).
Análisis de regresión lineal de las variables clínicas, bioquímicas y genéticas con la concentración de colesterol unido a lipoproteínas de baja densidad de familiares directos de personas con hipercolesterolemia genética sin hipercolesterolemia familiar
| Variable | Coeficiente β | IC95% | p | R2 corregido |
|---|---|---|---|---|
| Todos los familiares directos (n=268) | ||||
| Edad | 1,879 | 1,479-2,279 | <0,001 | 0,246 |
| Puntuación génica de colesterol | 0,576 | 0,220-0,932 | 0,002 | 0,277 |
| Perímetro de cintura | –0,558 | –1,090 a –0,027 | 0,040 | 0,286 |
| Familiares directos con puntuación génica de colesterol> media en el probando (n=136) | ||||
| Edad | 1,693 | 1,209-2,178 | <0,001 | 0,257 |
| Puntuación génica de colesterol | 0,857 | 0,401-1,313 | <0,001 | 0,326 |
IC95%: intervalo de confianza del 95%.
Modelo de regresión lineal ajustado por índice de masa corporal, edad, sexo y perímetro de cintura.
El R2 corregido explica el porcentaje de variabilidad de la variable dependiente (concentración de colesterol unido a lipoproteínas de baja densidad) que se explicaba por las variables independientes incluidas en el modelo (edad, puntuación genética de colesterol, índice de masa corporal, sexo y perímetro de cintura).
La regresión logística binaria mostró que por cada 0,016 unidades de aumento de la puntuación genética, el riesgo de tener el cLDL> percentil 90 aumentaba en 1,017 veces (intervalo de confianza del 95%, 1,001-1,033), con independencia de los factores de confusión (edad, sexo e índice de masa corporal) y determinaba el 19,1% de su variabilidad (área bajo la curva, 0,726).
DISCUSIÓNLas concentraciones de cLDL se deben a la interacción de múltiples factores genéticos y ambientales; así pues, la hipercolesterolemia tiende a acumularse en algunas familias que tienen en común una base genética y ambiental predisponente, lo que imita un fenotipo monogénico19. Además, la interacción de ciertos factores genéticos y ambientales, en especial el sobrepeso y el consumo de una dieta rica en calorías, tiene un efecto exponencial en las concentraciones de lípidos, como ocurre en la hiperlipemia combinada familiar, que anteriormente se consideraba una enfermedad monogénica, pero que luego se ha comprobado que se trata de una enfermedad compleja con un componente poligénico20. La consecuencia es que el diagnóstico en ciertas familias con cLDL alto en varios de sus miembros no es fácil, y en muchos casos (entre un 20 y un 40% de los pacientes con un diagnóstico clínico de HF) no se detecta un defecto de un solo gen y su hipercolesterolemia se debe a causas poligénicas8. Se ha recomendado que la denominación «familiar» se reserve para los trastornos derivados de un solo gen21 y que, cuando esto no se pueda demostrar, el diagnóstico de HF sea equívoco tanto para el médico como para el paciente; así pues, la designación de HGSHF define mejor las características de este grupo de pacientes con hipercolesterolemia11.
En varios estudios de asociación de genoma completo se ha observado que al menos 100 loci se asocian con la concentración de cLDL en la población22,23 y que algunos individuos portadores de múltiples SNV que aumentan el cLDL tienen unas concentraciones de cLDL altas que semejan el fenotipo de la HF13. Nosotros hemos analizado, por primera vez, las SNV mejor validadas que se asocian con el cLDL en grupos de familias con HGSHF y los resultados obtenidos indican varios aspectos importantes. En primer lugar, estos resultados confirman otros previos en cuanto al agrupamiento de determinadas SNV en los participantes con un diagnóstico de HGSHF e indican por primera vez que estas familias concentran alelos predisponentes que llevan a un aumento del cLDL en comparación con la población general y explican parte de su fenotipo. Esto tiene un gran valor, pues indica el carácter poligénico conceptual de esta hipercolesterolemia, aunque la cantidad de cLDL que se explica por estos factores genéticos es escasa. En segundo lugar, el estudio indica que la inclusión de la lipoproteína(a) en la puntuación génica mejora sustancialmente el porcentaje de variación del cLDL que explican las SNV. Dado que la concentración de lipoproteína(a) se debe principalmente a factores genéticos24, creemos que debe incluirse en las puntuaciones que se emplean para identificar a los participantes con HGSHF. En tercer lugar, tal como se preveía, la contribución de los factores genéticos difiere en gran manera en las distintas familias, y ello indica la complejidad y la heterogeneidad de la base genética de estas formas de hipercolesterolemia y pone en duda la utilidad diagnóstica de una sola puntuación genética basada en un pequeño grupo de SNV, que puede ser útil para casos seleccionados, pero puede tener poca eficacia en otras circunstancias. Indudablemente, se está aún lejos de disponer de una puntuación eficaz que identifique correctamente esta población, y serán necesarios más estudios para identificar mejor los genes causales. Por último, y como cuestión más importante, la ausencia de una mutación causal y la presencia de una puntuación poligénica alta no deben limitar el examen de detección familiar en cascada. Sin embargo, dicho examen debe basarse en la información clínica más que en la genética8. Aunque en estas familias la hipercolesterolemia no es monogénica, muchos individuos tienen concentraciones de cLDL muy altas, que requieren una identificación temprana. El objetivo del examen de detección en cascada no es identificar a los participantes con determinadas mutaciones, sino a los individuos con alto riesgo como consecuencia de sus altas concentraciones de cLDL25; el estudio muestra que el examen familiar en cascada basado en el cLDL debe realizarse a pesar de la ausencia de un defecto monogénico.
LimitacionesEl presente estudio tiene las siguientes limitaciones: el bajo número de SNV, que tal vez no sea la más importante asociación con el cLDL en nuestra población; la extrapolación del colesterol unido a una partícula de lipoproteína(a) basada en una fórmula uniforme, a pesar de que el contenido pueda diferir entre los participantes en función de cada isoforma de la apolipoproteína(a); la ponderación de cada SNV utilizada para el cálculo de la puntuación fue la media existente en la población, y el efecto en cada familia y cada individuo puede diferir, en función de otros factores genéticos y ambientales desconocidos. Por otro lado, los puntos fuertes de este estudio son que se estudió en profundidad el fenotipo y el genotipo de todos los participantes; se los reclutó en un solo centro, lo cual reduce la variabilidad, y procedían de una población genéticamente homogénea. Además, se ha demostrado que la puntuación de 6 SNV utilizada en este estudio es igual de buena discriminando entre HF e hipercolesterolemia no familiar que otras puntuaciones con múltiples SNV13, ya que la inclusión de otras SNV adicionales tuvo muy escaso efecto en la puntuación génica y las variaciones del cLDL, pero no mejoró el diagnóstico10.
En conclusión, el estudio de las SNV y la lipoproteína(a) en las familias con criterios clínicos de HF y sin mutaciones en genes candidatos pone de manifiesto el carácter poligénico de la enfermedad. Sin embargo, la puntuación genética basada en 7 marcadores genéticos explica solo un pequeño porcentaje de la hipercolesterolemia, lo que limita su uso diagnóstico. El componente poligénico de la hipercolesterolemia en estas familias con HGSHF no debe descartar el examen de detección familiar basado en el cLDL, ya que son frecuentes los casos de hipercolesterolemia grave en otros familiares.
FINANCIACIÓNEste trabajo fue financiado por el Ministerio de Sanidad de España, FIS PI13/02507, FIS PI15/01983, RD12/0042/0055, CIBERCV (con el apoyo de subvenciones europeas) y la Fundación Cuenca Villoro.
CONFLICTO DE INTERESESNo se declara ninguno.
- –
Algunas formas de hipercolesterolemia clasificadas como HF tienen un origen poligénico. Anteriormente se habían descrito 6 SNV asociadas con el diagnóstico de hipercolesterolemia poligénica. El valor de una puntuación genética basada en estas SNV asociadas con la hipercolesterolemia en familias afectadas no se había estudiado antes.
- –
Este es el primer estudio en el que se analiza la variación genética asociada con la hipercolesterolemia poligénica en familias con diagnóstico clínico de HF.
- –
El estudio genético familiar confirma el carácter poligénico de este fenotipo.
- –
Sin embargo, no resulta útil clínicamente diferenciar a los participantes con hipercolesterolemia de los normolipémicos.
- –
El diagnóstico de una hipercolesterolemia poligénica no debe descartar la aplicación de un examen de detección en cascada en los familiares, puesto que estas familias concentran a miembros con hipercolesterolemia grave.
Los análisis genéticos se realizaron en el centro de secuenciación y genómica funcional de los Servicios Científico Técnicos de CIBA (IACS-Universidad de Zaragoza), Zaragoza, España. Los autores agradecen a Maclean S. Panshin su inestimable ayuda en la corrección del inglés.