
La displasia valvular cardiaca ligada al cromosoma X es una cardiopatía congénita rara específica del sexo masculino y caracterizada principalmente por una degeneración mixomatosa de las válvulas auriculoventriculares con consecuencias hemodinámicas variables. Se debe a defectos genéticos en la filamina A (codificada por FLNA), una proteína de unión a actina de expresión ubicua que regula la organización del citoesqueleto. La pérdida de función de la filamina A también se ha asociado con manifestaciones neurológicas y del tejido conectivo a menudo simultáneas, y aparentemente las mutaciones en la primera mitad del dominio Rod 1 expresan el fenotipo cardiaco completo. En esta familia de nueva descripción, se ha contribuido a las correlaciones genotipo-fenotipo previas con un enfoque multidisciplinario.
MétodosLa evaluación cardiológica, dismorfológica y genética de los miembros disponibles se complementó con estudios de la transcripción y de la inactivación del cromosoma X.
ResultadosLa nueva mutación de FLNA c.1066-3C>G cosegregaba con un fenotipo cardiaco aparentemente aislado y expresado en los varones, sin que hubiera un sesgo en el patrón de inactivación del cromosoma X en las mujeres portadoras. Esta variante resultó en una deleción dentro del marco de lectura de 8 residuos de aminoácidos cercanos a la región N-terminal de la proteína.
ConclusionesLa pérdida de función parcial y no sometida a impronta del dominio Rod 1 proximal de la filamina A parece ser el mecanismo patogénico de la displasia valvular cardiaca, expresada en algunos casos con manifestaciones extracardiacas.
Palabras clave
La cardiopatía multivalvular es la combinación de lesiones de estenosis o insuficiencia en 2 o más válvulas cardiacas. Es un trastorno clínico de gran prevalencia entre los pacientes con una valvulopatía subyacente, y afecta a aproximadamente un 20% de los pacientes con defectos valvulares congénitos y un 15% de los sometidos a cirugía valvular1. En los ensayos PARTNER, la incidencia de la insuficiencia mitral moderada o grave concomitante en los pacientes con estenosis aórtica grave fue de alrededor de un 20% y la de insuficiencia tricuspídea moderada o grave, del 27%%; sin embargo, muchas de estas valvulopatías cardiacas fueron también secundarias a alteraciones hemodinámicas, cardiopatía isquémica o disfunción ventricular1. La complejidad de determinar la etiología de la enfermedad multivalvular dificulta el diagnóstico y el tratamiento de los pacientes.
La displasia valvular cardiaca ligada al cromosoma X (DVCX, MIM 314400), también denominada degeneración mixomatosa polivalvular, es un trastorno minoritario causado por mutaciones del gen de la filamina A (FLNA). Estos defectos genéticos subyacen al temprano inicio de un deterioro mixomatoso progresivo de las válvulas mitral y tricúspide que da lugar a engrosamiento y disfunción valvulares, seguido de un remodelado secundario de las cámaras y, en última instancia, insuficiencia cardiaca. Hasta la fecha se han descrito tan solo unos pocos casos y familias con este trastorno genético, algunos cuyos diagnósticos fueron retrospectivos2.
Se presenta una familia de 5 miembros en los que se confirmó o sospechó la presencia de una DVCX debida a una mutación nueva, c.1066-3C>G, en el intrón 7 de FLNA. Esta variante causa una abolición del lugar aceptor canónico de corte y empalme, con lo que da lugar a una deleción de 8 aminoácidos dentro del marco de lectura de la parte proximal del dominio Rod 1. Así pues, esta mutación puede conllevar una pérdida parcial de la función de la filamina A, lo cual coincide con lo observado en la mayoría de las variantes asociadas con DVCX descritas hasta ahora. Se examinan los casos de DVCX previos y se plantean hipótesis respecto a sus diferentes fenotipos, en función del efecto funcional causado, las repercusiones de estas variantes y la topología de la filamina A.
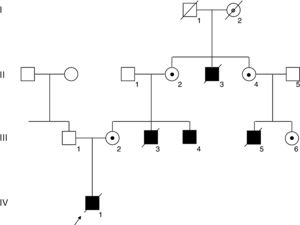
PacientesSe presenta un árbol genealógico completo de la familia en la figura 1.
. La mutación del gen FLNA se confirmó en el varón afectado vivo III-4 y las mujeres portadoras II-2, II-4, III-2 y III-6, y se indica con un punto negro. La portadora obligada I-2 se indica con un punto gris.")
Árbol genealógico de la familia. Los cuadrados y los círculos indican familiares de sexo masculino y femenino, respectivamente. Los familiares afectados se indican en negro y los fallecidos, con una raya cruzada. Una flecha indica al paciente índice (IV-1). La mutación del gen FLNA se confirmó en el varón afectado vivo III-4 y las mujeres portadoras II-2, II-4, III-2 y III-6, y se indica con un punto negro. La portadora obligada I-2 se indica con un punto gris.
Caso índice IV-1. Este paciente tenía diagnóstico prenatal de engrosamiento anormal de las válvulas cardiacas mediante ecografía y ecocardiografía fetal (vídeo 1 del material suplementario y vídeo 2 del material suplementario). Había una insuficiencia mitral moderada y una insuficiencia tricuspídea grave que causaba una dilatación aneurismática de la aurícula derecha. Las válvulas pulmonar y aórtica mostraban también una displasia y parecían pequeñas. El paciente nació a término (39 semanas), con un peso al nacer de 3.950g y una dificultad respiratoria neonatal grave que requirió oxigenación extracorpórea de membrana. En el primer día de vida, la ecocardiografía mostró una gran dilatación de la aurícula derecha, insuficiencia tricuspídea grave debida a una displasia valvular, insuficiencia mitral moderada con una válvula mitral displásica, una comunicación interauricular de 6mm con cortocircuito derecha-izquierda, dilatación del ventrículo derecho e hipertrofia ventricular izquierda. Las válvulas aórtica y pulmonar mostraban también displasia, con insuficiencia. La arteria pulmonar estaba moderadamente dilatada y el conducto arterioso era grande y tenía un flujo bidireccional, con predominio de izquierda-derecha. Había también un derrame pericárdico leve. La evolución clínica fue la propia de un paciente con un corazón izquierdo hipoplásico y atresia aórtica funcional y dependencia de un conducto arterioso permeable. A la exploración física no se describieron manifestaciones propias de una conectivopatía. Se le practicó una intervención de cirugía cardiaca para reparar ambas válvulas, pero el paciente falleció durante la intervención. Ni la autopsia ni la ecografía cerebral previa mostraban trastornos de migración neuronal. El cariotipo estándar era normal.
Familiar afectado III-4. Se trata de un varón de 27 años con una cardiopatía congénita. Nació a término con síndrome de dificultad respiratoria. A los 2 meses de edad, se le detectó un soplo cardiaco. La ecocardiografía posnatal confirmó que todas las válvulas mostraban engrosamiento y distrofia, con insuficiencia tricuspídea moderada e insuficiencias mitral y pulmonar leves, así como una leve incompetencia aórtica (vídeo 3 del material suplementario). La exploración clínica no mostró hipermovilidad articular ni hiperextensiblidad cutánea. La resonancia magnética cerebral no detectó ningún trastorno de migración neuronal.
Familiar afectado III-3. Este familiar nació a término tras un embarazo y un parto aparentemente normales. Poco después del nacimiento, el paciente presentó cianosis y dificultad respiratoria y falleció en el primer día de vida, hace más de 30 años. No se dispone de más datos.
Familiar afectado III-5. Este niño nació de un parto pretérmino (37 semanas) después de un embarazo y un parto normales. Permaneció en la unidad de cuidados intensivos neonatales durante 2 semanas a causa de la prematuridad y la alimentación anormal. Se le practicó una intervención de cirugía cardiaca en otro país y, después de 2 meses de un curso clínico indolente, falleció a causa de una enfermedad respiratoria. La familia indicó que el recién nacido tenía anomalías valvulares cardiacas con dilatación de cavidades y un corazón grande.
Familiar afectado II-3. Este niño nació de un parto a término en el domicilio hace más de 60 años. La única información aportada por la familia es que el niño tenía soplos cardiacos y un retraso del crecimiento. No se dispone de más datos.
Se realizó una ecocardiografía a las mujeres emparentadas asintomáticas (III-2, II-2, II-4, III-6), con resultados normales en todos los casos excepto el de una mujer de 58 años (familiar II-2) que presentaba una insuficiencia pulmonar leve. A la exploración clínica, no se observó hipermovilidad articular ni hiperextensiblidad cutánea. No se realizaron exploraciones por ecografía o resonancia magnética cerebral.
MÉTODOSAnálisis genéticoTodos los participantes dieron su consentimiento informado para los estudios realizados y se obtuvo la aprobación ética de los respectivos centros. Se realizó una amplificación y secuenciación de la secuencia codificante de FLNA y de los límites intrón-exón (NM_001456.3) mediante ADN genómico extraído de linfocitos de sangre periférica de la madre del probando (III-2), ya que no se pudo obtener ninguna muestra del caso índice. Posteriormente se realizaron estudios de segregación familiar.
Estudios del ARNSe obtuvieron muestras de sangre periférica del varón afectado vivo (III-4) en tubos de sangre para ARN PAXgene (PreAnalytiX GmBH, Qiagen NV/Becton Dickenson & company) para la extracción de ARN. El ARN se extrajo con el kit de ARN en sangre (PreAnalytiX GmBH) y se realizó una síntesis del ADN complementario (ADNc) con el High Capacity cDNA Reverse Transcription Kit (ThermoFisher Scientific), en ambos casos según las instrucciones del fabricante. El ADNc se amplificó con un amplicón que contiene los exones 6-9 de FLNA (NM_001456.3), utilizando los oligonucleótidos: 5’-ACACCAGGAGGAGGCAAAAG-3’, situado en la unión de los exones 6 y 7, y 5’-GCTCTACCGTGCCCTTCTGT-3’, situado en el exón 9. Los productos se visualizaron mediante electroforesis y luego se secuenciaron.
Inactivación del cromosoma XEl patrón de inactivación del cromosoma X se determinó con el ADN genómico procedente de los linfocitos de las mujeres portadoras disponibles mediante un test cuantitativo de metilación indirecto en el gen del receptor de andrógenos, según un método descrito con anterioridad3.
Análisis bioinformáticoLas variantes con una frecuencia del alelo minoritario inferior al 0,1% se filtraron según los datos disponibles en la base de datos gnomAD4. Se determinaron las predicciones de patogenicidad in silico con el programa Alamut Visual V2.9.0, que incluye las siguientes herramientas de simulación de corte y empalme: SpliceSiteFinder-like, MaxEntScan, NNSPLICE, GeneSplicer y Human Splicing Finder. La clasificación de las variantes se realizó según las normas y la guía de la ACMG para la interpretación de las variantes de secuencia5.
RESULTADOSSe detectó una variante intrónica heterocigota, c.1066-3C>G, en el gen FLNA de la madre del caso índice (III-2, figura 2A), en la posición -3 del lugar aceptor de corte y empalme del intrón 7. Esta variante estaba ausente en más de 177.600 alelos de la base de datos gnomAD. Los familiares II-2, II-4, III-4 y III-6 presentaron también resultados positivos para la misma variante, lo cual confirmó la segregación en todos los familiares afectados y portadores disponibles e indicó que el familiar I-2 era un portador obligado (figura 1).
, una portadora homocigota (III-2, panel central) y un varón afectado hemicigoto (III-4, panel inferior); se indica el límite entre el intrón 7 y el exón 8. B: predicciones de corte y empalme realizadas in silico que muestran la consecuencia de la variante en el lugar aceptor del intrón 7; obsérvese que existe un lugar aceptor críptico fuerte localizado a 24 pb distalmente en el exón 8. C: secuenciación del ADNc en el paciente III-4 que muestra la abolición real del lugar aceptor de corte y empalme canónico en el intrón 7 en el ADNc mutante y la utilización del lugar aceptor distal alternativo en el exón 8; esto da lugar a una deleción en el marco de lectura de 24 pb (8 aminoácidos). Esta figura se muestra a todo color solo en la versión electrónica del artículo.")
Estudios genéticos. A: cromatograma de secuenciación genómica que muestra la variante c.1066-3C>G en un control normal (WT, panel superior), una portadora homocigota (III-2, panel central) y un varón afectado hemicigoto (III-4, panel inferior); se indica el límite entre el intrón 7 y el exón 8. B: predicciones de corte y empalme realizadas in silico que muestran la consecuencia de la variante en el lugar aceptor del intrón 7; obsérvese que existe un lugar aceptor críptico fuerte localizado a 24 pb distalmente en el exón 8. C: secuenciación del ADNc en el paciente III-4 que muestra la abolición real del lugar aceptor de corte y empalme canónico en el intrón 7 en el ADNc mutante y la utilización del lugar aceptor distal alternativo en el exón 8; esto da lugar a una deleción en el marco de lectura de 24 pb (8 aminoácidos). Esta figura se muestra a todo color solo en la versión electrónica del artículo.
El aceptor del lugar de corte y empalme canónico en el intrón 7 estaba totalmente inactivado en la secuencia mutada, según SpliceSiteFinder-like, MaxEntScan y GeneSplicer, y estaba significativamente debilitado (76,17 frente a 86,47) según lo predicho por Human Splicing Finder (figura 2B). Según las predicciones de corte y empalme in silico, existe un lugar aceptor críptico en una posición 24 pb distal dentro del exón 8, que puede dar lugar al rescate del marco de lectura de la proteína (p.Val356_Gln363del). Esta predicción se confirmó en los estudios de ARN de la sangre periférica del paciente III-4 (figura 2C).
El patrón de inactivación del cromosoma X se determinó para las portadoras III-2, II-2 y III-6. Todas ellas mostraron un patrón aleatorio de inactivación (50:50, 45:55 y 44:56, respectivamente). La portadora II-4 no fue informativa respecto al polimorfismo utilizado en esta prueba.
DISCUSIÓNSe han descrito formas sindrómicas y formas aisladas de las valvulopatías hereditarias. Las conectivopatías, como el síndrome de Marfan y, más raramente, el síndrome de Loeys-Dietz, muestran prolapso de la válvula mitral y otras disfunciones valvulares que forman parte de un espectro clínico vascular, ocular y óseo más amplio6. El síndrome de Ehlers-Danlos, especialmente la forma valvular cardiaca autosómica recesiva, causada por mutaciones del gen COL1A2, también tiene manifestaciones valvulares izquierdas de diversos niveles de gravedad7.
La DVCX fue la primera distrofia valvular no sindrómica que se caracterizó desde el punto de vista genético mediante un análisis de ligamiento, que mostró defectos de la filamina A como etiología subyacente8. Se diferencia por una disfunción multivalvular bilateral de inicio precoz que afecta predominantemente al lado izquierdo, probablemente por el mayor estrés hemodinámico de las válvulas mitral y áortica2. Se hereda en forma de rasgo ligado al cromosoma X, de un modo aparentemente recesivo que muestra habitualmente una penetrancia completa en los varones y una expresión subclínica reducida en las mujeres portadoras8.
Sin embargo, las mutaciones de FLNA se han asociado también con una amplia variedad de enfermedades genéticas diferentes, entre las que se encuentran: la heterotopia nodular periventricular (HNPV, MIM 300049)9, el síndrome FG de tipo 2 (MIM 300321), la seudooclusión intestinal neuronal (MIM 300048), la displasia ósea terminal (MIM 300244) y los trastornos del espectro otopalatodigital de tipo I (MIM 311300) y II (MIM 304120), el síndrome de Melnick-Needles (MIM 309350) y la displasia frontometafisaria (MIM 305620)10. Se ha propuesto que la pérdida total o parcial de la función de la filamina A da lugar a trastornos de la migración neuronal y distrofias valvulares respectivamente, mientras que las displasias óseas parecen estar en relación con un mecanismo de ganancia de función10. No obstante, se han observado excepciones a este modelo en cuanto a los resultados fenotípicos, la expresión tisular y la letalidad esperada en los varones11–13. Se han descrito también fenotipos mixtos14, incluidos los efectos funcionales opuestos, dependientes de transcriptos, de variantes únicas15,16.
Ocasionalmente se han observado defectos graves de la válvula mitral o la válvula pulmonar en pacientes con HNPV17,18. Antes de que se publicaran estas observaciones, la válvula aórtica bicúspide, la dilatación de la raíz aórtica y el conducto arterioso permeable eran las únicas manifestaciones cardiovasculares que se habían asociado con la HNPV. Además, se ha hallado también un fenotipo similar al del síndrome de Ehlers-Danlos, que incluye hipermovilidad articular e hiperlaxitud cutánea en pacientes con una HNPV asociada con el gen FLNA19–21. Tanto los defectos valvulares como las conectivopatías forman parte de un mismo espectro etiológico relacionado con la matriz extracelular, y de hecho ambos trastornos se han observado de manera simultánea en pacientes con filamina A22–25. A diferencia de las displasias óseas, todos estos defectos parecen tener relación con una pérdida completa o parcial de la función de la filamina A y pueden formar parte de un mismo espectro clínico continuo. La DVCX relacionada con el gen FLNA no debe considerarse una forma no sindrómica de la distrofia valvular, sino más bien un trastorno pleotrópico de expresividad variable, posiblemente relacionada con la alteración funcional y las repercusiones de la mutación.
De hecho, en los casos de DVCX descritos anteriormente, las manifestaciones del tejido conjuntivo e incluso la HNPV parecen ser frecuentes en los pacientes en los que se examinaron estas características (tabla). Las manifestaciones óseas marfanoides se evalúan sistemáticamente cuando se identifican defectos valvulares congénitos, mientras que la HNPV, incluso en ausencia de epilepsia o crisis convulsivas, tan solo puede descartarse mediante resonancia magnética cerebral. Por otra parte, las únicas familias que parecen mostrar HNPV tienen mutaciones de FLNA no especificadas que no revelan el dominio afectado o las repercusiones de la mutación (tabla).
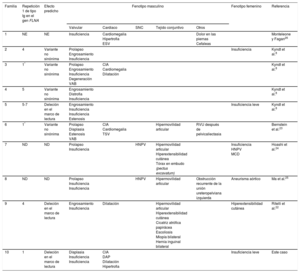
Resumen de los fenotipos cardiacos y extracardiacos y de los resultados genéticos en los pacientes con displasia valvular cardiaca ligada al cromosoma X
| Familia | Repetición 1 de tipo Ig en el gen FLNA | Efecto predicho | Fenotipo masculino | Fenotipo femenino | Referencia | ||||
|---|---|---|---|---|---|---|---|---|---|
| Valvular | Cardiaco | SNC | Tejido conjuntivo | Otros | |||||
| 1 | NE | NE | Insuficiencia | Cardiomegalia Hipertrofia ESV | Dolor en las piernas Cefaleas | Monteleone y Fagan26 | |||
| 2 | 4 | Variante no sinónima | Prolapso Engrosamiento Insuficiencia | Insuficiencia | Kyndt et al.8 | ||||
| 3 | 1* | Variante no sinónima | Prolapso Engrosamiento Insuficiencia Degeneración VAB | CIA Cardiomegalia Dilatación | Kyndt et al.8 | ||||
| 4 | 5 | Variante no sinónima | Engrosamiento Distrofia Insuficiencia | Kyndt et al.8 | |||||
| 5 | 5-7 | Deleción en el marco de lectura | Engrosamiento Insuficiencia Insuficiencia Estenosis | Insuficiencia leve | Kyndt et al.8 | ||||
| 6 | 1* | Variante no sinónima | Prolapso Displasia Estenosis VAB | CIA Cardiomegalia TSV | Hipermovilidad articular | RVU después de pelvicaliectasia | Bernstein et al.23 | ||
| 7 | ND | ND | Prolapso Insuficiencia | HNPV | Hipermovilidad articular Hiperextensibilidad cutánea Tórax en embudo (pectus excavatum) | Insuficiencia HNPV MCD | Hoashi et al.24 | ||
| 8 | ND | ND | Prolapso Insuficiencia Insuficiencia | HNPV | Hipermovilidad articular | Obstrucción recurrente de la unión ureteropelviana izquierda | Aneurisma aórtico | Ma et al.25 | |
| 9 | 4 | Deleción en el marco de lectura | Engrosamiento Insuficiencia | Dilatación | Hipermovilidad articular Hiperextensibilidad cutánea Cicatriz atrófica papirácea Escoliosis Miopía bilateral Hernia inguinal bilateral | Hiperextensibilidad cutánea | Ritelli et al.22 | ||
| 10 | 1 | Deleción en el marco de lectura | Displasia Insuficiencia Insuficiencia | CIA DAP Dilatación Hipertrofia | Insuficiencia leve | Este caso | |||
CIA: comunicación interauricular; DAP: conducto arterioso permeable; ESV: extrasístoles ventriculares; HNPV: heterotopia nodular periventricular; MCD: miocardiopatía dilatada; ND: no disponible; NE: no evaluado; RVU: reflujo vesicoureteral; SNC: sistema nervioso central; TSV: taquicardia supraventricular; VAB: válvula aórtica bicúspide.
No obstante, los individuos descritos en este estudio no presentan manifestaciones extracardiacas, neurológicas, óseas o cutáneas. El paciente índice nació con una comunicación interauricular, malformación esta para la que anteriormente se ha descrito una asociación con defectos de la filamina A. Es de destacar que los otros 2 pacientes con DVCX y este defecto cardiaco eran portadores de la misma mutación, también en la repetición 1 de tipo Ig del dominio Rod 1 (tabla). Los defectos vasculares presentes en el paciente índice se han descrito también en pacientes con HNPV y, en general, los defectos cardiovasculares son frecuentes en las displasias óseas asociadas con el gen FLNA14.
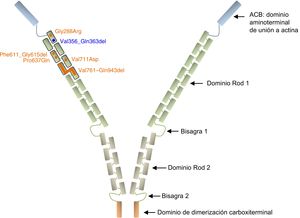
La filamina A es una proteína ubicua que estabiliza los filamentos de actina y los une a glucoproteínas de membrana. También se ha descrito que interactúa con otras muchas proteínas, como las integrinas, los complejos de receptores transmembranarios y los segundos mensajeros, lo cual indica un papel en la transducción de señal. Estructuralmente, el dominio de unión a la actina conservado en el extremo aminoterminal de la secuencia proteica va seguido de 24 repeticiones de tipo Ig homólogas de 96 aminoácidos cada una. Esta estructura principal está separada por 2 dominios de bisagra, que separan el Rod 1 (repeticiones 1-15) del Rod 2 (repeticiones 16-23), que se siguen de un dominio de dimerización carboxiterminal (repetición 24)27,28 (figura 3).

Representación esquemática de la proteína filamina A. Las variantes no sinónimas se representan en forma de círculos y las deleciones intragénicas, como barras. Todos los cambios descritos en la DVCX se indican en naranja, excepto el descrito en este artículo, que se muestra en azul. Obsérvese que todas las alteraciones están agrupadas en las primeras 7 repeticiones de tipo Ig del dominio Rod 1 de la proteína. Las referencias relativas a cada variante se citan en el texto. Esta figura se muestra a todo color solo en la versión electrónica del artículo.
De las 9 familias con DVCX descritas con anterioridad, se observaron mutaciones de FLNA en 8 (los pacientes de la familia descrita por Monteleone y Fagan en 1969 no fueron evaluados al respecto)26. En 2 de las 8 familias positivas para FLNA, no se ha especificado la mutación/cambio de proteína24,25, mientras que en las 6 restantes todas las variantes estaban situadas en las repeticiones de Ig 1 a 7, dentro del dominio Rod 1 (p.Gly288Arg, c.1829-1G>C, p.Pro637Gln, p.Val711Asp, p.Val761_Gln943del)8,22,23,29 (figura 3). Todas estas variantes probablemente actúen como alelos hipomórficos que mantienen el marco de lectura de un dominio presumiblemente no crítico, por lo que pueden conservar cierta actividad residual de la proteína, puesto que se han observado en varones hemicigotos vivos. Para la variante c.1829-1G>C, se predijo una abolición del lugar aceptor de corte y empalme del intrón 12, lo cual indica una activación posterior de un lugar aceptor críptico situado a 15 pb distalmente en el exón 13, lo cual conlleva una deleción en el marco de lectura de 5 aminoácidos consecutivos (p.Phe611_Gly615del)22. Es de destacar que una variante con el mismo efecto predicho de corte y empalme se ha asociado anteriormente con HNPV y disfunción valvular mitral leve18.
Se predijo también que la variante detectada en la familia destruiría el lugar aceptor de corte y empalme canónico del intrón 7, con el uso de un lugar alternativo situado a 24 pb distalmente en el exón 8. En este caso, la alteración de corte y empalme se confirmó en los estudios del ARN y causó una deleción en el marco de lectura de 8 aminoácidos conservados (p.Val356_Gln363del). Las variantes que conservan el marco de lectura situadas en la parte proximal del dominio Rod 1 del gen FLNA se consideran asociadas con un fenotipo cardiaco aislado en los varones afectados. Sin embargo, las manifestaciones extracardiacas observadas en estos pacientes apuntan más bien a un espectro continuo entre las variantes con pérdida parcial y pérdida total de función (tabla).
Los lugares aceptores crípticos, situados distalmente a los lugares aceptores de corte y empalme canónicos, intervienen en el mecanismo molecular de muchos trastornos humanos. Se ha observado el uso de estos lugares alternativos para el rescate del marco de lectura en otros genes en los que una pérdida parcial de la función puede tolerarse mejor, como COL1A1 y COL1A2 en el síndrome de Ehlers-Danlos de tipo VII en comparación con la osteogénesis imperfecta30, COL5A1 en el síndrome de Ehlers-Danlos de los tipos I y II31, DMD en las distrofias musculares de Duchenne y Becker32 y LAMA2 en la distrofia de cinturas autosómica recesiva en comparación con la distrofia muscular congénita33. Sin embargo, no se puede explicar por qué los pacientes con la deleción de 5 aminoácidos en la repetición 4 de FLNA expresaban las manifestaciones extracardiacas tan graves, observadas al parecer incluso en la paciente antecesora, mientras que la deleción del exón 4 con falta de las repeticiones 5 a 7 y la deleción actual de 8 aminoácidos en la repetición 1 parecen estar limitadas a un fenotipo cardiaco aislado. Es posible que haya interacciones diferentes, dependientes de la molécula de unión en estas regiones, así como otros factores genéticos, que originen los fenotipos observados.
Se ha propuesto también una asimetría en la inactivación del cromosoma X en la modulación de la expresión fenotípica en las mujeres portadoras de variantes patógenas del gen FLNA. Sin embargo, una asimetría extrema de la inactivación del cromosoma X hacia el alelo mutado solo se ha observado de manera persistente en las displasias óseas con afectación sistémica congénita asociadas con el gen FLNA, lo cual concuerda con un requisito celular de función de la filamina A durante el desarrollo10. No obstante, en las alteraciones de pérdida de función no se ha descrito ningún patrón específico que se asocie con el grado en que la enfermedad se manifiesta9. Se observó un patrón de inactivación del cromosoma X aleatorio en 3 mujeres portadoras, todas ellas asintomáticas y 1 de ellas con una disfunción valvular subclínica leve. Así pues, no parece haber una correlación clínica entre la expresión fenotípica y el patrón de inactivación del cromosoma X, lo cual respalda las observaciones previas. A pesar de ello, debe señalarse que el proceso de inactivación del cromosoma X puede ser dependiente del tejido y que su determinación en el ADN genómico procedente de linfocitos puede no ser aplicable a los tejidos afectados.
Se sabe que la filamina A se une a varias proteínas de unión además de los filamentos de actina, lo cual probablemente explique la diversidad de los fenotipos resultantes descrita. Se cree que la expresión fenotípica valvular y posiblemente otras manifestaciones en los tejidos blandos de los trastornos asociados con el gen FLNA tiene relación con la interacción de la filamina A con los componentes de la vía de señalización del TGF-β, a la que se ha involucrado ya en conectivopatías que producen aneurismas de la raíz aórtica y prolapso de la válvula mitral34. Sin embargo, la mayoría de las proteínas con interacción identificadas hasta la fecha no se unen a la región aminoterminal del dominio Rod 1, donde se ha descrito que están las mutaciones asociadas con la DVCX. Recientemente se ha caracterizado funcionalmente el papel patógeno de estas mutaciones en la reducción de las capacidades de migración y extensión celular por medio de un deterioro de la red de señalización de la Rho-GTPasa que modifica las vías de remodelado de la actina, que son importantes para las respuestas celulares ante el estrés mecánico, las interacciones de las células con la matriz extracelular y la transformación epitelial-mesenquimatosa35.
CONCLUSIONESLos resultados obtenidos respaldan que las variantes que conservan el marco de lectura en las repeticiones 1-7 del dominio Rod 1 de la filamina A tienen algún papel en la expresión de un fenotipo principal de la DVCX, que asimismo puede incluir manifestaciones de expresión variable en el tejido conjuntivo y posiblemente también neurológicas. La observación de un fenotipo de DVCX aparentemente aislado debe motivar la búsqueda de estas manifestaciones relacionadas más allá de los trastornos del colágeno y la fibrilina36. Identificar las alteraciones genéticas en FLNA en estos pacientes confirma su diagnóstico, mejora el pronóstico y facilita el asesoramiento genético a las familias.
FINANCIACIÓNEste trabajo fue financiado en parte por la subvención PI13/1450 del ISCII (Instituto de Salud Carlos III) y la subvención SAF2015-66831-R del MINECO (Ministerio de Economía, Industria y Competitividad) y cofinanciado por el FEDER (Fondo Europeo de Desarrollo Regional).
CONFLICTO DE INTERESESNo se declara ninguno.
- –
La DVCX es un trastorno congénito minoritario que se expresa predominantemente en los varones. La causan mutaciones de FLNA, que es el gen que codifica la filamina A, una proteína intermediaria entre el citoesqueleto y la membrana. A diferencia de otras formas autosómicas de valvulopatías, que incluyen conectivopatías, puede producirse de manera aislada, si bien se ha observado cierto solapamiento entre diferentes fenotipos relacionados con la filamina A. Se cree que los diferentes dominios, efectos de las mutaciones, proteínas de interacción y factores genéticos intervienen en esta variabilidad.
- –
Se ha observado una nueva mutación en el gen FLNA en una familia de 3 generaciones con DVCX que se ha descrito clínicamente. Los estudios del ARN en el varón afectado confirmaron una deleción en el marco de lectura en la parte proximal del dominio Rod 1 y, por lo tanto, una pérdida parcial de la función proteica como mecanismo de la enfermedad predicho. Se han examinado los casos de DVCX descritos anteriormente, teniendo en cuenta la localización predicha de la mutación y el daño proteico cuando se conocen, y se han evaluado los fenotipos extracardiacos. Se propone un espectro continuo de todos los fenotipos asociados con pérdida de función de la filamina A, lo cual indica la necesidad de examinarlos en los pacientes con valvulopatías cardiacas.
Los autores desean expresar su agradecimiento a todos los pacientes y familiares por su amable apoyo.