Una función endocrina normal es esencial para la salud cardiovascular. Los trastornos del sistema endocrino, consistentes en hiperfunción o hipofunción hormonales, tienen múltiples efectos en el sistema cardiovascular. En esta revisión se comentan la epidemiología, el diagnóstico y el tratamiento de los trastornos de las glándulas hipófisis, tiroides, paratiroides y suprarrenales en lo relativo a sus repercusiones de disfunción endocrina en el sistema cardiovascular. Se revisan también los efectos beneficiosos cardiovasculares que aporta el restablecimiento de una función endocrina normal.
Palabras clave
Una función endocrina normal es esencial para la salud cardiovascular. Los trastornos del sistema endocrino, consistentes en hiperfunción o hipofunción hormonales, tienen múltiples efectos en el sistema cardiovascular. El objetivo de esta revisión es explorar los diversos cambios cardiovasculares que se producen en la disfunción endocrina. Se evalúan también los efectos beneficiosos cardiovasculares de la corrección de los trastornos endocrinos. Se excluye específicamente la diabetes, ya que la relación bien conocida entre diabetes y riesgo cardiovascular queda fuera del ámbito de esta revisión.
La hipófisis y el sistema cardiovascularVisión general de la hipófisisLa hipófisis anterior contiene cinco tipos de células que sintetizan y secretan hormonas (hormona de crecimiento [GH], prolactina, folitropina, lutropina, tirotropina y corticotropina [ACTH]) que participan en la regulación del eje hipotálamo-hipófisis-órgano diana. La hipófisis posterior contiene terminaciones nerviosas que secretan vasopresina (hormona antidiurética) y oxitocina. De las hormonas hipofisarias secretadas por la hipófisis anterior, los trastornos de la prolactina, la GH y la ACTH pueden asociarse a cardiopatías.
Trastornos de la prolactina y enfermedad cardiovascularLa prolactina se sintetiza y se secreta por las células lactotrofas de la hipófisis anterior y estimula la lactación en el periodo posparto. La dopamina hipotalámica inhibe tónicamente la prolactina. La concentración de prolactina está elevada fisiológicamente en el embarazo, el periodo posparto y los estados de estrés. La hiperprolactinemia patológica puede tener causa en una disminución de la inhibición dopaminérgica, como ocurre cuando se produce una sección del tallo hipofisario, o porque haya secreción de prolactina por prolactinomas (adenomas hipofisarios benignos). La prevalencia de hiperprolactinemia oscila entre el 0,4% en la población general adulta y el 9% en mujeres con trastornos de la reproducción1. Aunque la hiperprolactinemia en sí no tiene efectos claros en el sistema cardiovascular, hay una posible asociación entre el tratamiento dopaminérgico a largo plazo y las anomalías valvulares cardiacas.
Los dopaminérgicos, como cabergolina, bromocriptina y quinagolida (no autorizada en Estados Unidos), constituyen el tratamiento primario de los prolactinomas. La cabergolina es la más utilizada, dadas su eficacia clínica, su tolerabilidad y su perfil farmacocinético favorable2. Las dosis altas y la duración prolongada del tratamiento con dopaminérgicos en la enfermedad de Parkinson se han asociado a un aumento del riesgo de insuficiencia valvular cardiaca3,4. Aunque las dosis utilizadas en el tratamiento del prolactinoma son muy inferiores a las que se usan para la enfermedad de Parkinson, los pacientes con prolactinomas pueden ser tratados durante décadas. Esta duración del tratamiento plantea la preocupación de un posible aumento del riesgo de valvulopatía, incluidas la insuficiencia tricuspídea, la insuficiencia mitral y la insuficiencia aórtica5,6. Aunque la mayoría de los estudios no muestran una asociación entre el uso de dopaminérgicos y las valvulopatías cardiacas, se recomienda a los clínicos que utilicen las dosis de dopaminérgicos lo más bajas posible. Debe considerarse la posible conveniencia de un seguimiento ecocardiográfico de los pacientes que necesitan un tratamiento a largo plazo o dosis más altas, así como de los que presenten una cardiopatía o valvulopatía subyacente7.
La miocardiopatía periparto es una entidad clínica muy poco frecuente. Se ha señalado que un fragmento de prolactina de 16 kDa puede participar en su fisiopatología8. Se han descrito casos de uso de bromocriptina además de un tratamiento estándar para la insuficiencia cardiaca en la miocardiopatía periparto9.
Visión general de la hormona de crecimientoLa GH se sintetizada y se secretada por las células somatotrofas en la hipófisis anterior. La GH actúa directamente sobre los tejidos periféricos a través de una interacción con el receptor de GH, e indirectamente por estimulación de la síntesis del factor insulinoide de crecimiento tipo 1 (IGF-1). El IGF-1 fomenta la captación de glucosa y la síntesis de proteínas celulares en prácticamente todos los tipos de células. La GH y el IGF-1 regulan el crecimiento somático, incluidos el desarrollo y la función cardiacos10.
La prevalencia del déficit de GH (DGH) en los adultos es de aproximadamente 1-2/10.00011. La prevalencia de la acromegalia, o exceso de secreción de GH, es de aproximadamente 40-70 casos/millón, con una incidencia estimada de 3-4/millón al año12,13.
Visión general del déficit de hormona de crecimientoLos adultos con DGH pueden agruparse en tres categorías: los que presentan un DGH de inicio en la infancia, los que tienen un DGH adquirido, secundario a lesiones estructurales o traumáticas, y los que presentan un DGH idiopático de inicio en el adulto14. El diagnóstico se confirma por valores bajos de IGF-1 en suero y mediante pruebas de provocación mediante hipoglucemia inducida por insulina y la combinación de arginina y somatoliberina (GHRH), que son estímulos potentes para la secreción de GH. Un aumento inferior al normal de la concentración sérica de GH tras la prueba de tolerancia a la insulina o la prueba de GHRH-arginina confirma el diagnóstico de DGH15. El tratamiento del DGH consiste en una terapia sustitutiva de GH.
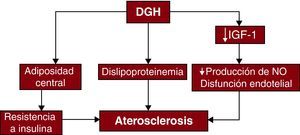
Déficit de hormona de crecimiento y enfermedad cardiovascularRiesgo cardiovascularEl DGH se asocia a un aumento de la grasa corporal y la adiposidad central, dislipemia (colesterol unido a lipoproteínas de alta densidad [cHDL] bajo, colesterol total elevado y colesterol unido a lipoproteínas de baja densidad [cLDL] alto), disfunción endotelial y resistencia a la insulina16,17 (Figura 1). Se ha descrito también en el DGH un aumento del grosor de la íntima-media (GIM) de la arteria carótida, que es un marcador del desarrollo aterosclerótico temprano19,20. El tratamiento sustitutivo de GH puede producir un aumento de la masa corporal magra y una disminución del tejido adiposo visceral21. La reposición de GH puede reducir la concentración de colesterol total y cLDL, pero los efectos en el cHDL han sido inconstantes22. La disfunción endotelial mejora con la terapia sustitutiva de GH, con un aumento de la dilatación mediada por flujo y una reducción de la rigidez arterial a causa de la mejora de disponibilidad de óxido nítrico (NO)23. Aunque se ha demostrado que la terapia sustitutiva de GH reduce el GIM, los efectos en la evolución cardiovascular son inciertos24.

Figura 1. Efecto del déficit de hormona de crecimiento en la aterosclerosis. DGH: déficit de hormona de crecimiento; IGF-1: factor insulinoide de crecimiento tipo 1; NO: óxido nítrico. Adaptada con permiso de Colao 18 .
Estructura y función cardiacasLa ecocardiografía en pacientes con un DGH de inicio en la infancia o en la adolescencia ha mostrado reducciones significativas del grosor de la pared posterior del ventrículo izquierdo (VI) y del grosor del tabique interventricular, con la consiguiente reducción del índice de masa y el diámetro interno del VI25,26. La mayor parte de los pacientes adultos con DGH presentan un deterioro de la función del VI en ejercicio máximo y refieren intolerancia al esfuerzo27. Varios estudios han mostrado que la terapia sustitutiva de GH mejora la función cardiaca y aumenta la masa del VI, el volumen telediastólico del VI (VTDVI) y el volumen de eyección25,28.
Visión general de la acromegaliaLa acromegalia se caracteriza por volumen circulante elevado de GH y IGF-1 y la causa es un adenoma hipofisario benigno en más del 98% de los casos. La morbimortalidad asociada a la acromegalia se debe a los efectos metabólicos de la hipersecreción de GH/IGF-1 y los efectos de masa del adenoma hipofisario. La media de edad en el momento del diagnóstico es de 40-45 años, habitualmente con un periodo de 5-10 años de síntomas antes de que se establezca el diagnóstico. Los síntomas consisten en disminución de la tolerancia al ejercicio, aumento del tamaño del anillo o del ajuste de este en el dedo, aumento del número de calzado, prominencia de la mandíbula o la frente, acné o piel grasienta, artropatías y neuropatías29,30.
El diagnóstico de la acromegalia se sospecha por valores elevados de IGF-1 y se confirma al determinar una elevación de la GH tras una prueba de sobrecarga oral de glucosa. Los tratamientos para la acromegalia se dirigen a reducir o controlar el crecimiento del adenoma, inhibir la hipersecreción de GH y normalizar los valores de IGF-I. La cirugía es el tratamiento de primera línea para la acromegalia. Las opciones de tratamiento para los valores persistentemente elevados de GH y/o IGF-1 son el tratamiento médico y la radioterapia. Las tres clases de fármacos existentes para el tratamiento de la acromegalia son los análogos de somatostatina, los agonistas dopaminérgicos y los antagonistas de receptores de GH31.
Acromegalia y enfermedad cardiovascularRiesgo cardiovascularSe produce hipertensión en un 20-50% de los pacientes con acromegalia. Los posibles mecanismos son el aumento de la rigidez arterial a causa de hipertrofia y fibrosis de la túnica muscular arterial32. La acromegalia se asocia también a un aumento de la prevalencia de diabetes mellitus33. La presión arterial sistólica y diastólica y el control de la glucemia mejoran con la normalización de los valores de IGF-134.
Estructura y función cardiacasLas anomalías histológicas cardiacas en la acromegalia incluyen hipertrofia miocitaria, fibrosis intersticial, infiltración de células inflamatorias, reducción de la densidad capilar, alteración miofibrilar y depósito de colágeno extracelular. Las repercusiones de estas alteraciones en la estructura y la función de los tejidos miocárdicos y vasculares vienen dadas por la duración y la gravedad del exceso de GH/IGF-1. En el estadio inicial de la acromegalia, se producen aumento de la contractilidad miocárdica, reducción de la resistencia vascular sistémica, aumento del gasto cardiaco y aumento general de la función cardiaca. El grosor relativo de la pared (grosor de la pared del VI/radio del VI) aumenta, y ello causa una reducción de la tensión en la pared. En el estadio intermedio, tras unos 5 años de enfermedad activa, se producen hipertrofia biventricular, disfunción diastólica y deterioro de la función cardiaca en esfuerzo. La miocardiopatía acromegálica en estadio avanzado se caracteriza por disfunción sistólica y diastólica, aumento de la masa miocárdica, dilatación de la cavidad ventricular y aumento de la resistencia vascular sistémica32. La miocardiopatía acromegálica con frecuencia está presente en el momento del diagnóstico. Hasta dos tercios de los pacientes con acromegalia cumplen los criterios ecocardiográficos de HVI, incluida aproximadamente la mitad del total de pacientes con acromegalia normotensos. Los pacientes con miocardiopatía grave pueden presentar progresión a la insuficiencia cardiaca, de tal manera que esta se observa en un 3-10% de los pacientes35. El tratamiento eficaz de la acromegalia detiene la progresión de la disfunción cardiaca y reduce la mortalidad cardiovascular29. Se ha descrito que la curación quirúrgica reduce la masa cardiaca y mejora el llenado diastólico36. Se ha demostrado que el control satisfactorio de la enfermedad con análogos de somatostatina mejora los parámetros de llenado diastólico, reduce la sobrecarga de volumen, reduce las presiones pulmonar y enclavada y aumenta la función cardiaca37. Hay alguna evidencia de que la hipertrofia cardiaca es reversible en los pacientes jóvenes con corto tiempo de evolución de la enfermedad27. Se observa también mejoría de la fracción de eyección del VI en ejercicio máximo en los pacientes jóvenes con corto tiempo de evolución de la enfermedad38.
Las valvulopatías cardiacas (insuficiencia aórtica y mitral) son frecuentes en la acromegalia39. El exceso de GH/IGF-1 puede conducir a una regulación anormal de la matriz extracelular y, por lo tanto, a la patogenia de la valvulopatía mixomatosa. El riesgo de valvulopatía aumenta significativamente con la duración del exceso de GH. La disfunción valvular aórtica y mitral persiste a menudo a pesar del tratamiento del exceso hormonal40.
RitmoLos exámenes de electrocardiograma (ECG) y Holter han documentado anomalías del ritmo cardiaco en la acromegalia. Las alteraciones observadas en el ECG en reposo incluyen la desviación del eje a la izquierda, el aumento de los intervalos QT, las ondas Q septales y la depresión de la onda ST-T41. Además, hasta un 56% de los pacientes con acromegalia activa presentan potenciales tardíos en el ECG que podrían predisponer a las arritmias42. Las alteraciones del ritmo se observan principalmente durante el ejercicio físico, y entre ellas hay extrasístoles auriculares y ventriculares, fibrilación auricular paroxística, taquicardia supraventricular paroxística, síndrome del seno enfermo, bloqueo de rama del haz y taquicardia ventricular. La frecuencia de las extrasístoles ventriculares aumenta con el mayor tiempo de evolución de la acromegalia. La gravedad de las arritmias ventriculares se correlaciona con los aumentos de la masa ventricular izquierda43. Se ha demostrado que los análogos de somatostatina reducen los intervalos QT y mejoran el perfil arrítmico en los pacientes acromegálicos44.
Visión general de la corticotropinaLa ACTH se sintetiza y se secreta por las células corticotropas de la hipófisis anterior. La función principal de la ACTH es regular la secreción suprarrenal de cortisol. El exceso de ACTH puede producirse por un adenoma corticotropo hipofisario o, excepcionalmente, por un tumor extrahipofisario (síndrome de ACTH ectópica), como un cáncer de pulmón microcítico, tumor carcinoide o cáncer medular de tiroides. Este exceso de secreción de ACTH produce hipercortisolismo o síndrome de Cushing. El síndrome de Cushing endógeno se produce por secreción excesiva de ACTH (dependiente de ACTH) en aproximadamente el 80% de los casos, y por causas independientes de la ACTH en aproximadamente un 20% de los casos, entre los que se encuentran la secreción de cortisol por adenomas suprarrenales unilaterales o por una hiperplasia o displasia suprarrenal bilateral45. La incidencia global de síndrome de Cushing endógeno es de 2,3 casos/millón al año46.
El diagnóstico del síndrome de Cushing exige la demostración de títulos elevados de cortisol con al menos dos pruebas de confirmación, que pueden ser el cortisol libre en orina de 24 h, el cortisol libre salival nocturno tardío o la prueba de supresión con dexametasona durante una noche47,48. Los objetivos del tratamiento en el síndrome de Cushing son la normalización y el control a largo plazo de los valores de cortisol y la reversión de las manifestaciones clínicas como aumento de peso, obesidad central, fatiga, debilidad muscular, hipertensión, diabetes, hirsutismo, acné y trastornos menstruales. Las opciones de tratamiento incluyen la cirugía transesfenoidal, la suprarrenalectomía unilateral o bilateral, la radioterapia y el tratamiento médico. La elección y la eficacia de una determinada modalidad de tratamiento dependen de la causa subyacente del hipercortisolismo49. El control médico del hipercortisolismo en los pacientes no quirúrgicos puede alcanzarse con el empleo de ketoconazol, metirapona o mitotano50.
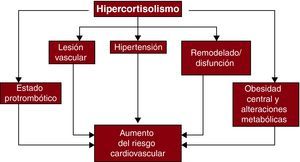
Síndrome de Cushing y enfermedad cardiovascularRiesgo cardiovascularEl hipercortisolismo causa hipertensión, obesidad central, resistencia a la insulina, dislipemia y alteraciones de la coagulación y de la función plaquetaria51 (Figura 2). Alrededor del 80% de los pacientes adultos con síndrome de Cushing endógeno tienen hipertensión, que se debe a alteraciones de la regulación del volumen plasmático, resistencia vascular sistémica y vasodilatación53,54. El tratamiento del síndrome de Cushing suele comportar mejoría o resolución de la hipertensión, aunque esta puede persistir en los pacientes con un hipercortisolismo de larga evolución y/o una hipertensión esencial coexistente55. El metabolismo anormal de la glucosa en el síndrome de Cushing se debe a la estimulación de la neoglucogénesis y la glucogenólisis hepáticas. Los pacientes con hipercortisolismo pueden presentar intolerancia a la glucosa en ayunas, hiperinsulinemia, resistencia a la insulina y diabetes mellitus56. El síndrome de Cushing se ha asociado a aumento de las concentraciones de lipoproteína (a), disminución del cHDL y aumento de los triglicéridos54. La duración del exceso de cortisol se correlaciona con el grado de dislipemia observado. El cortisol aumenta también la síntesis de varios factores de la coagulación, estimula la producción endotelial de factor von Willebrand y eleva al mismo tiempo el factor VIII57. Además, el hipercortisolismo puede potenciar la agregación plaquetaria y reducir la capacidad fibrinolítica del plasma58,59.

Figura 2. Mecanismos de aumento del riesgo cardiovascular a través del hipercortisolismo. Reproducida con permiso de Fallo et al 52 .
Estructura y función cardiacasEl síndrome de Cushing se ha asociado a HVI, remodelado concéntrico, disfunción diastólica y disfunción sistólica del VI subclínica60. La ecocardiografía ha revelado un aumento del grosor del tabique interventricular y del grosor de la pared posterior, un aumento del índice de masa del VI y un aumento del grosor relativo de la pared en los pacientes con síndrome de Cushing. Se ha demostrado una disfunción diastólica con deterioro de la relajación inicial del VI, aumento de los tiempos de relajación isovolumétrica y signos de deterioro de la relajación general del miocardio. Las anomalías de la estructura y la función del VI pueden ser reversibles con la normalización del hipercortisolismo. Sin embargo, los pacientes pueden continuar presentando intolerancia al esfuerzo a causa de la miopatía inducida por los corticoides y la consiguiente debilidad muscular61.
Tiroides y sistema cardiovascularVisión general del tiroidesLa disfunción tiroidea es frecuente. El hipertiroidismo se da en un 1,3% de la población de Estados Unidos (manifiesto en un 0,5% y subclínico en un 0,7%), y el hipotiroidismo afecta a un 4,6% de la población (manifiesto en un 0,3% y subclínico en un 4,3%)62. La prevalencia tanto del hipotiroidismo como del hipertiroidismo aumenta con la edad. Los datos del Framingham Heart Study han puesto de relieve supresión de los niveles de tirotropina (TSH) en el 3,9% de los pacientes de más de 60 años y cierto grado de hipotiroidismo, indicado por una cifra de TSH sérica elevada (> 5 mU/l) en el 10,3% de los pacientes no seleccionados de más de 60 años de edad, con mayor incidencia en las mujeres (13,6%) que en los varones (5,7%)63,64.
Visión general del hipertiroidismoLa tirotoxicosis manifiesta o hipertiroidismo se define por una elevación de las concentraciones de hormonas tiroideas libres en sangre periférica (T3 y/o T4) y una TSH disminuida o indetectable. La tirotoxicosis puede ser consecuencia de una enfermedad autoinmunitaria, autonomía de un nódulo tiroideo o el aporte exógeno de hormonas tiroideas. Los pacientes hipertiroideos a menudo presentan signos y síntomas relacionados con el sistema cardiovascular, como palpitaciones, taquicardia sinusal, fibrilación auricular, hipertensión sistólica, presión del pulso ensanchada, intolerancia al esfuerzo y disnea de esfuerzo. Otros síntomas consisten en fatiga, pérdida de peso, intolerancia al calor y diarrea. Los tratamientos disponibles para el hipertiroidismo incluyen las medicaciones antitiroideas (metimazol, carbimazol y propiltiouracilo), los bloqueadores beta, la ablación con yodo radiactivo y la cirugía tiroidea. El hipertiroidismo subclínico se define como una concentración sérica baja o indetectable de TSH junto con cifras normales de hormona tiroidea libre periférica. Los pacientes suelen estar asintomáticos; no obstante, tienen riesgo de sufrir algunas alteraciones cardiovasculares asociadas al hipertiroidismo65. Las recomendaciones de un panel de consenso señalan que se considere el tratamiento en caso de déficit persistente de la TSH sérica (TSH<0,1 UI/l)66.
Hipertiroidismo y enfermedad cardiovascularHemodinámicaLas acciones genómicas y no genómicas de la hormona tiroidea dan lugar a alteraciones hemodinámicas cardiovasculares en el hipertiroidismo manifiesto, que consisten en disminución de la resistencia vascular sistémica (RVS), aumento de la frecuencia cardiaca, aumento de la precarga cardiaca y aumento del gasto cardiaco67,68. La RVS se reduce en el hipertiroidismo a causa de la relajación de las células de músculo liso vascular mediada por la hormona tiroidea y el aumento de la producción de NO endotelial69,70. La disminución de la RVS activa el sistema renina-angiotensina-aldosterona, lo que da lugar a un aumento del volumen plasmático y un incremento de la precarga cardiaca. La hormona tiroidea fomenta también un aumento del volumen sanguíneo a través de regulación al alza de la secreción de eritropoyetina, lo cual incrementa aún más la precarga cardiaca71. La combinación de aumento de la precarga y disminución de la RVS conduce a elevación del gasto cardiaco72. Los aumentos de la contractilidad y la frecuencia cardiacas en reposo contribuyen también a elevar el gasto cardiaco, que puede ser un 50-300% superior al normal en los pacientes con hipertiroidismo manifiesto73,74. El tratamiento del hipertiroidismo revierte estas alteraciones hemodinámicas.
Riesgo cardiovascularPuede observarse hipertensión sistólica en hasta un 30% de los pacientes hipertiroideos75. Esta elevación de la presión puede deberse al efecto combinado del aumento de la precarga y el gasto cardiaco y la disminución de la distensibilidad arterial76.
Estructura y función cardiacasSe ha asociado HVI al hipertiroidismo77. Las alteraciones hemodinámicas que se producen en el hipertiroidismo causan un aumento del trabajo cardiaco y una hipertrofia cardiaca compensatoria al cabo del tiempo78. El hipertiroidismo se asocia también a un aumento de la relajación diastólica. A corto plazo, el hipertiroidismo puede asociarse a una mejora de la función diastólica, pero a largo plazo, la tirotoxicosis crónica puede inducir HVI y disfunción diastólica79. La intolerancia al ejercicio y la disnea de esfuerzo en el hipertiroidismo manifiesto pueden deberse a la incapacidad de incrementar en mayor medida la frecuencia cardiaca y la fracción de eyección o a que se reduce más la RVS cuando se realiza ejercicio. Los pacientes con hipertiroidismo pueden tener también debilidad del músculo esquelético o respiratorio que reduzca todavía más la capacidad de ejercicio. Los pacientes con hipertiroidismo subclínico pueden presentar reducción de la tolerancia al ejercicio65. El tratamiento del hipertiroidismo produce una mejoría de la tolerancia al ejercicio y resuelve la disnea de esfuerzo80.
RitmoLa taquicardia sinusal se produce en aproximadamente un 40% de los casos de hipertiroidismo manifiesto, y generalmente se resuelve tras el restablecimiento del eutiroidismo81. El hipertiroidismo subclínico se asocia también a un aumento de la frecuencia cardiaca65. La fibrilación auricular es la segunda arritmia más frecuente en el hipertiroidismo manifiesto, y se produce en un 10-15% de los pacientes; su prevalencia aumenta con la edad82. Los pacientes con hipertiroidismo subclínico presentan un aumento del riesgo de fibrilación auricular65,83. En los casos de hipertiroidismo manifiesto, los factores independientes que predicen la fibrilación auricular son la edad creciente, los antecedentes de insuficiencia cardiaca, la diabetes, la presión arterial sistólica o diastólica elevada y la HVI en el ECG84. Puede restablecerse el ritmo sinusal en hasta dos tercios de los pacientes con hipertiroidismo manifiesto, pero la mayor edad y el mayor tiempo de evolución de la fibrilación auricular se asocian a tasas más altas de arritmia persistente84. Hay escasa evidencia de que el tratamiento del hipertiroidismo subclínico facilite la reversión de la fibrilación auricular a ritmo sinusal normal66.
Visión general del hipotiroidismoLos pacientes con hipotiroidismo pueden presentar fatiga, aumento de peso, intolerancia al frío, estreñimiento, hipertensión diastólica leve, estrechamiento de la presión del pulso y bradicardia. El hipotiroidismo manifiesto se caracteriza por una elevación de la TSH sérica y una disminución de los valores de hormona tiroidea en sangre periférica, y su etiología incluye el fallo autoinmunitario de la glándula tiroides, el fallo iatrogénico (yodo radiactivo, radioterapia de haz externo) o la tiroidectomía. El tratamiento del hipotiroidismo consiste en una terapia sustitutiva de tiroxina (T4). El hipotiroidismo subclínico se define por una TSH sérica elevada con valores de hormona tiroidea libre periférica normales. Los pacientes con hipotiroidismo subclínico suelen estar asintomáticos o levemente sintomáticos. Las recomendaciones de un panel de consenso indican la instauración de una terapia sustitutiva de hormona tiroidea en los pacientes con valores de TSH sérica>10 mUI/l y proponen que se considere la posible conveniencia de una terapia sustitutiva en los pacientes con una TSH sérica de 4,5-10 mUI/l que presentan síntomas, un riesgo cardiovascular basal elevado y/o autoinmunidad tiroidea85.
Hipotiroidismo y enfermedad cardiovascularHemodinámicaLas alteraciones hemodinámicas que se producen en el hipotiroidismo son opuestas a las observadas en el hipertiroidismo. El hipotiroidismo manifiesto se asocia a aumento de la RVS, frecuencia cardiaca en reposo normal o reducida, disminución de la contractilidad y descenso del gasto cardiaco. Además, hay un aumento de la presión diastólica y se produce un estrechamiento de la presión del pulso. El gasto cardiaco puede estar reducido en hasta un 30-40% como consecuencia de la reducción del volumen de eyección y la frecuencia cardiaca86. Las alteraciones hemodinámicas que se dan en el hipotiroidismo se resuelven cuando se restablece el eutiroidismo, con normalización de la RVS y mejoría de la contractilidad cardiaca y el gasto cardiaco87.
Riesgo cardiovascularEl hipotiroidismo manifiesto se asocia a aterosclerosis acelerada y enfermedad coronaria, que pueden ser atribuibles a la hipertensión diastólica, el deterioro de la función endotelial y la hipercolesterolemia. Puede observarse hipertensión diastólica significativa en hasta un 20% de los pacientes con hipotiroidismo manifiesto. Este aumento de la presión diastólica es consecuencia de un aumento de la resistencia vascular sistémica y una mayor rigidez arterial, y se resuelve con terapia sustitutiva de T488. El hipotiroidismo manifiesto se ha asociado también a hiperhomocisteinemia, aumento de proteína C reactiva y alteración de los parámetros de la coagulación88. El hipotiroidismo subclínico se ha asociado a elevación de la presión diastólica y aumento del GIM carotídeo, que puede mejorar con la terapia sustitutiva de T465.
El metabolismo lipídico está alterado en el hipotiroidismo, y aproximadamente un 90% de los pacientes con hipotiroidismo manifiesto presentan unas concentraciones elevadas de colesterol total y cLDL89. Las concentraciones séricas de colesterol total y cLDL están aumentadas en aproximadamente un 30% en el hipotiroidismo, y los aumentos observados de las LDL son mayores en los pacientes con resistencia a la insulina y en los fumadores. Los valores de LDL están aumentados principalmente a causa de la disminución de la eliminación fraccional de LDL que se produce como consecuencia de la reducción en el número de receptores de LDL hepáticos. La apolipoproteína B y la variante de LDL aterogénica, la lipoproteína (a), están aumentadas también en el hipotiroidismo. Los triglicéridos y las lipoproteínas de muy baja densidad (VLDL) son normales o están aumentados, mientras que los cambios de las HDL tienen un carácter variable90. Estas anomalías lipídicas generalmente son reversibles al restablecer el eutiroidismo. El hipotiroidismo subclínico se ha asociado a aumento de cLDL y colesterol total en varios estudios transversales, pero los efectos del tratamiento en ensayos de pequeño tamaño han sido poco concordantes65.
Estructura y función cardiacasEn el hipotiroidismo, hay disfunción diastólica ventricular izquierda en reposo y disfunción tanto sistólica como diastólica con el ejercicio. En el hipotiroidismo manifiesto, se ha demostrado un deterioro de la función diastólica ventricular izquierda por la relajación miocárdica más lenta y el deterioro del llenado ventricular temprano88. En los pacientes ancianos que pueden presentar una mayor rigidez miocárdica preexistente, el hipotiroidismo manifiesto puede conducir a insuficiencia cardiaca diastólica. La terapia sustitutiva de T4 resuelve estas anomalías funcionales y mejora la función tanto sistólica como diastólica. También se han demostrado modificaciones de la disfunción diastólica del VI en reposo en los pacientes con hipotiroidismo subclínico, con mejora en respuesta a la terapia sustitutiva de T465. Se producen derrames pericárdicos en hasta un 25% de los pacientes con hipotiroidismo manifiesto, y es probable que se deban a aumento de la permeabilidad capilar, aumento del volumen de distribución de la albúmina y deterioro del drenaje linfático67. Estos derrames pericárdicos se acumulan lentamente y rara vez tienen trascendencia hemodinámica, aunque se han descrito casos poco comunes de taponamiento cardiaco91. Los derrames pericárdicos asociados al hipotiroidismo se resuelven generalmente después de 2-3 meses de terapia sustitutiva de hormona tiroidea67.
RitmoLas alteraciones del ECG en el hipotiroidismo incluyen bradicardia sinusal, complejos de bajo voltaje (ondas P o complejos QRS pequeños), intervalos PR o QT prolongados y aplanamiento o inversión de las ondas T92. Se han descrito casos de anomalías de la conducción ventricular de forma asociada al hipotiroidismo, y es posible que esto esté relacionado con la prolongación del intervalo QT73.
Amiodarona y hormona tiroideaLa amiodarona, un fármaco antiarrítmico benzofuránico rico en yodo, causa disfunción tiroidea en un 15-20% de los pacientes tratados, y ello conduce a hipotiroidismo o tirotoxicosis. El hipotiroidismo inducido por amiodarona (HIA) se debe a la inhibición persistente de la función de la glándula tiroides inducida por el yodo y es más prevalente en los pacientes con autoinmunidad tiroidea preexistente93. El tratamiento del HIA se realiza con sustitución de T4. A menudo son necesarias dosis altas de T4, ya que la amiodarona reduce la actividad de desyodasa, lo que causa disminución de la conversión de T4 a la forma activa, T3. La tirotoxicosis inducida por amiodarona (TIA) se manifiesta en dos formas: TIA tipo 1 o hipertiroidismo inducido por yodo y TIA tipo 2 o tiroiditis destructiva. La TIA tipo 1 da lugar a la síntesis y liberación de un exceso de hormona tiroidea, mientras que la TIA tipo 2 causa la liberación de la hormona tiroidea preformada por parte de la glándula tiroides inflamada. Diferenciar las dos formas y el tratamiento de la TIA pueden resultar difíciles. La TIA tipo 1 se trata con fármacos antitiroideos y posiblemente con perclorato potásico. La TIA tipo 2 se trata con glucocorticoides, bloqueadores beta y, excepcionalmente, tiroidectomía94 (Tabla 1). Deben realizarse pruebas basales de la función tiroidea y determinaciones de anticuerpos contra la peroxidasa tiroidea antes de iniciar el tratamiento con amiodarona, y debe efectuarse seguimiento de la función tiroidea cada 6 meses mientras se mantenga dicho tratamiento95.
Tabla 1. Características de la disfunción tiroidea inducida por amiodarona.
| Tirotoxicosis tipo I | Tirotoxicosis tipo II | Hipotiroidismo | |
| Mecanismo | Exceso de yodo. Más frecuente en áreas con déficit de yodo | Tiroiditis inflamatoria destructiva | Exceso de yodo. Más frecuente en áreas con suficiencia de yodo |
| Anticuerpos tiroideos | Presentes con frecuencia | Habitualmente negativos | Presentes con frecuencia |
| Función tiroidea | Tirotoxicosis | Tirotoxicosis | Hipotiroidismo |
| Captación de 123I durante 24 h | Generalmente baja en las regiones con suficiencia de yodo, pero puede ser normal o estar aumentada en las áreas con déficit de yodo | < 5% | Generalmente baja en las áreas con suficiencia de yodo |
| Ecografía Doppler color | Hipervascularidad | Reducción del flujo sanguíneo | Vascularidad normal |
| Tratamiento | Dosis altas de fármacos antitiroideos; posiblemente perclorato o ácido iopanoico antes de la tiroidectomía | Corticoides en dosis altas; ácido iopanoico | Levotiroxina sódica |
Reproducida con permiso de Pearce et al 95 .
Una concentración sérica baja de T3 es la anomalía de la función tiroidea que se observa con mayor frecuencia en los pacientes con insuficiencia cardiaca, y se da en alrededor de un 10-30% de los pacientes96. El perfil bioquímico de la función tiroidea en la insuficiencia cardiaca concuerda con el de la enfermedad no tiroidea o el del síndrome de enfermo eutiroideo. Continúa sin estar claro si esta reducción de la T3 es un proceso adaptativo o constituye una mala adaptación97. Tampoco está claro el papel de la terapia sustitutiva tiroidea en los pacientes con insuficiencia cardiaca y cifras de T3 bajas. Los objetivos del tratamiento incluirán mejoría en la función del VI, el remodelado y la microcirculación. Los campos de investigación actuales al respecto incluyen la terapia sustitutiva tiroidea con T3 y/o T4, el uso de análogos de hormona tiroidea (p. ej., ácido diyodotiropropiónico) y la terapia génica para modificar la expresión y la actividad de desyodasa o del receptor de hormona tiroidea98. Sin embargo, estos enfoques por el momento son experimentales.
Paratirina y sistema cardiovascularVisión general de la hormona paratiroideaLa hormona paratiroidea (PTH) tiene un papel crucial en el mantenimiento de una homeostasis calcio-fósforo adecuada99. La PTH afecta a tres órganos diana principales en el mantenimiento del equilibrio del calcio: hueso, mucosa intestinal y riñones. La incidencia de hiperparatiroidismo primario (HPTP) es de aproximadamente 21,6/100.000 al año, con mayor incidencia en mujeres y adultos de edad avanzada, de tal manera que se alcanza un máximo de 63,2/100.000 al año a la edad de 65-74 años100. El hipoparatiroidismo es mucho menos frecuente.
Visión general del hiperparatiroidismoEl hiperparatiroidismo se caracteriza por una elevación inadecuada de los títulos de PTH en el contexto de unas concentraciones de calcio elevadas. Las causas de hiperparatiroidismo incluyen el HPTP debido a un adenoma autónomo o una hiperplasia de la glándula paratiroides, y el hiperparatiroidismo secundario debido a enfermedad renal crónica o déficit de vitamina D de larga evolución. La forma de presentación clínica del HPTP ha evolucionado en los últimos años con la mejora de la detección de la enfermedad. Aproximadamente un 85% de los pacientes que presentan HPTP están asintomáticos o tienen sólo síntomas mínimos. El diagnóstico del hiperparatiroidismo se realiza mediante la determinación de las concentraciones séricas de calcio y PTH intacta y observando valores de PTH inadecuadamente altos en presencia de valores elevados de calcio. A los pacientes con un hiperparatiroidismo se les debe ofrecer un tratamiento quirúrgico con extirpación del adenoma o los adenomas paratiroideos hiperfuncionantes. En los pacientes asintomáticos, puede optarse por seguimiento clínico o se les puede ofrecer el tratamiento quirúrgico si cumplen los criterios clínicos apropiados101.
Hiperparatiroidismo y enfermedad cardiovascularRiesgo cardiovascularEl riesgo cardiovascular asociado al HPTP es atribuible en gran parte a un aumento de la prevalencia de hipertensión, obesidad, intolerancia a la glucosa y resistencia a la insulina102,103. Los mecanismos de hipertensión propuestos en los pacientes con HPTP incluyen el aumento del depósito de calcio que conduce a una rigidez arterial en la enfermedad de larga duración o grave, la estimulación directa del sistema renina-aldosterona a través de la PTH y la disfunción endotelial a través de la PTH, con aumento de la actividad simpática104,105. La corrección quirúrgica del hiperparatiroidismo no ha mostrado de manera uniforme una mejoría de la hipertensión106,107. Se ha demostrado que el tratamiento del HPTP con cirugía mejora la sensibilidad a la insulina en los pacientes con una enfermedad más grave108,109. Se ha observado que el GIM carotídeo es mayor en los pacientes con HPTP, y las medidas de la rigidez carotídea se asocian al grado de elevación de la PTH. Esto indica que la rigidez vascular puede estar relacionada con la gravedad del hiperparatiroidismo110.
Estructura y función cardiacasSe ha observado HVI en el HPTP en muchos estudios, en especial en pacientes con hiperparatiroidismo de moderado a grave, con independencia de los efectos de la hipertensión. Los datos de estudios realizados en animales indican que la PTH tiene efectos tróficos en los miocardiocitos que causan una hipertrofia. La corrección quirúrgica del hiperparatiroidismo ha conseguido regresión de la HVI en algunos estudios111.
Se ha documentado disfunción diastólica en el HPTP de moderado a grave, y se ha descrito una reducción del cociente E/A con una prolongación del tiempo de relajación isovolumétrico. Sin embargo, continúa sin estar claro si este efecto es atribuible en mayor medida a la hipercalcemia o al exceso de PTH111. El HPTP se ha asociado de manera poco constante a anomalías de la disfunción diastólica.
Se han demostrado calcificaciones de la válvula aórtica, la válvula mitral y el miocardio en los pacientes con HPTP e hipercalcemia significativa102. Sin embargo, los estudios realizados con hipercalcemia de leve a moderada no han mostrado una correlación consistente con el aumento de las calcificaciones valvulares112.
RitmoLa hipercalcemia, y en particular un valor de calcio sérico > 12 mg/dl, reduce la fase de meseta del potencial de acción cardiaco ventricular y el periodo refractario efectivo. Los signos electrocardiográficos en la hipercalcemia significativa incluyen acortamiento de los intervalos QT y QTc, aumento de la amplitud del complejo QRS, el máximo temprano y la pendiente negativa gradual de la rama descendente de la onda T, ondas T bifásicas y acortamiento del segmento ST92. Una corrección quirúrgica satisfactoria del hiperparatiroidismo, con reducción de las concentraciones séricas de calcio, puede comportar la prolongación de los intervalos QT y QTc113. Continúa sin estar claro si el hiperparatiroidismo y la hipercalcemia dan lugar a anomalías de la conducción cardiaca clínicamente relevantes114,115.
Visión general del hipoparatiroidismoEl hipoparatiroidismo se caracteriza por valores inadecuadamente bajos o indetectables de PTH en el contexto de una hipocalcemia. El hipoparatiroidismo puede ser congénito o adquirido, y la causa adquirida más frecuente es la lesión o extirpación quirúrgica de las glándulas paratiroides116. Los signos y síntomas del hipoparatiroidismo son consecuencia de la hipocalcemia. La hipocalcemia leve puede manifestarse por irritabilidad neuromuscular, como entumecimiento peribucal, calambres musculares, parestesias y signos de Chvostek y Trousseau positivos. La hipocalcemia grave puede manifestarse por espasmos carpopedales, laringospasmo, tetania y crisis epilépticas. El examen diagnóstico debe incluir determinaciones de las concentraciones séricas de calcio total y ionizado, albúmina, fósforo, magnesio, creatinina, PTH intacta y 25-hidroxivitamina D117. El tratamiento consiste en una terapia sustitutiva adecuada de calcio mediante la administración de calcio oral o i.v., y metabolitos y análogos de la vitamina D según sea necesario.
Hipoparatiroidismo y enfermedad cardiovascularEstructura y función cardiacasSe han descrito casos de disminución de la función miocárdica, miocardiopatía dilatada e insuficiencia cardiaca congestiva en pacientes con hipocalcemia aguda o crónica118,119. El mecanismo de la disfunción miocárdica no está claro, pero puede estar relacionado con un deterioro del acoplamiento de excitación-contracción. Se ha observado reversión de la insuficiencia cardiaca y corrección de la miocardiopatía en algunos casos en que fue necesaria la corrección del déficit de calcio para obtener una mejoría clínica y hemodinámica120,121.
RitmoLa prolongación del QT es la marca electrocardiográfica distintiva de la hipocalcemia y se debe a una prolongación de la fase de meseta del potencial de acción ventricular. La rapidez del cambio de la concentración de calcio extracelular modula la función de los canales del calcio. Los cambios rápidos del calcio sérico comportan cambios más notables del intervalo QT122. Los cambios de la onda T no son frecuentes en la hipocalcemia, ya que la fase 3 del potencial de acción no se ve afectada. Sin embargo, en la hipocalcemia grave, se ha descrito aplanamiento de la onda T, inversión de la onda T terminal u ondas T invertidas y profundas92. La hipocalcemia se ha asociado también, infrecuentemente, a elevación del segmento ST, posiblemente a causa de un espasmo arterial coronario123.
La glándula suprarrenal y el sistema cardiovascularVisión general de la aldosteronaLa aldosterona es una hormona mineralocorticoidea producida en la glándula suprarrenal. La secreción de aldosterona se regula fundamentalmente por el sistema renina-angiotensina, aunque otros factores reguladores que intervienen en ello son la concentración sérica de sodio y potasio y la ACTH. Las hormonas mineralocorticoideas mantienen las concentraciones normales de sodio y potasio, así como el estado de volumen normal.
Visión general del aldosteronismo primarioEl aldosteronismo primario (AP), o hiperaldosteronismo primario, es un grupo de trastornos en los que la producción de aldosterona es inadecuadamente alta, con lo que se produce una supresión del sistema renina-angiotensina. La hipertensión es la característica distintiva del AP; se ha descrito una prevalencia de AP de un 0,5-4,8% en los pacientes con hipertensión general y de un 4,5-22% en los pacientes con hipertensión refractaria124. La depleción de potasio también es característica del hiperaldosteronismo. El diagnóstico del AP se hace inicialmente determinando la aldosterona plasmática y la actividad de renina en plasma y calculando la proporción de aldosterona respecto a renina (ARR). En los pacientes con una ARR positiva (ARR > 20 con aldosterona > 15 ng/dl), debe realizarse una prueba de confirmación (carga de sodio oral, infusión de suero fisiológico, supresión de fludrocortisona o exposición a captopril). Las causas frecuentes de AP son el adenoma suprarrenal autónomo unilateral y la hiperplasia suprarrenal unilateral o bilateral. Una causa infrecuente de AP es un trastorno hereditario denominado aldosteronismo remediable con glucocorticoides (ARG). Las guías de tratamiento recomiendan una suprarrenalectomía laparoscópica unilateral en los pacientes con AP unilateral documentado o el tratamiento médico con un antagonista de los receptores de mineralocorticoides (espironolactona o eplerenona) en los pacientes no quirúrgicos125. Se indica tratamiento médico a los pacientes con enfermedad suprarrenal bilateral. Se indica terapia sustitutiva de glucocorticoides a las dosis terapéuticas más bajas posibles como tratamiento para el ARG.
Aldosteronismo primario y enfermedad cardiovascularRiesgo cardiovascularEl AP se asocia a hipertensión, disfunción endovascular y alteración del metabolismo de la glucosa. Los mecanismos que contribuyen a producir la hipertensión en el hiperaldosteronismo son la expansión del volumen plasmático a causa de la retención de sodio y líquidos y la vasoconstricción por depleción de potasio126. Se ha demostrado que la aldosterona reduce la biodisponibilidad de NO e inhibe la relajación dependiente del endotelio. La fibrosis perivascular que se produce a través de la aldosterona reduce la distensibilidad vascular124. La suprarrenalectomía laparoscópica bilateral en pacientes con adenomas productores de aldosterona o hiperplasia suprarrenal unilateral normaliza la hipopotasemia en todos los pacientes, mejora el control de la presión arterial en casi todos y obtiene tasas hipertensión resuelta a largo plazo de un 30-60%. En el AP debido a enfermedad suprarrenal bilateral, la suprarrenalectomía unilateral o bilateral rara vez corrige la hipertensión, y se hace necesario continuar con el tratamiento con antagonistas de los receptores de mineralocorticoides127. Se ha descrito intolerancia a la glucosa y reducción de la sensibilidad a la insulina en algunos pacientes con AP. Los mecanismos propuestos son los efectos directos de la aldosterona en la función del receptor de insulina y los efectos de la hipopotasemia en la regulación de la insulina128.
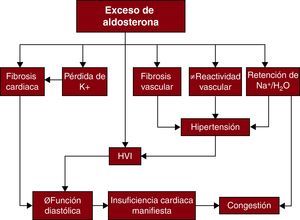
Estructura y función cardiacasEl hiperaldosteronismo causa un remodelado cardiaco mal adaptado y se ha asociado a HVI, fibrosis cardiaca y disfunción diastólica129,130,131 (Figura 3). El grado de HVI que se observa en el AP supera el que se explica por el efecto de la hipertensión por sí sola132. En modelos animales, se ha demostrado que la aldosterona estimula directamente el crecimiento celular y la hipertrofia de los miocardiocitos133. También se ha observado que la aldosterona fomenta el depósito de colágeno, la activación de las células inflamatorias y la estimulación de la proliferación fibroblástica134,135. Se ha demostrado una disfunción diastólica con un valor más bajo de la relación de velocidades de llenado diastólico de onda inicial/tardía y un tiempo de desaceleración superior en los pacientes con AP136. Los tratamientos médicos y quirúrgicos pueden ser efectivos para reducir la masa del VI, y las reducciones de la presión arterial y las concentraciones de aldosterona en plasma predicen la respuesta al tratamiento.

Figura 3. Mecanismos por los que el exceso de aldosterona puede comportar secuelas cardiovasculares adversas. HVI: hipertrofia ventricular izquierda. Adaptada con permiso de Stowasser 131 .
Insuficiencia cardiaca congestiva y bloqueo de aldosteronaEn trastornos como la insuficiencia cardiaca y el infarto de miocardio, los valores de aldosterona están aumentados y contribuyen a causar el remodelado cardiovascular patológico a través de sus efectos directos en el depósito de colágeno y la consiguiente fibrosis cardiovascular137. Los valores elevados de aldosterona fomentan también la disfunción endotelial y la inflamación vascular. En estudios clínicos se ha demostrado que el bloqueo de la aldosterona reduce el remodelado del VI y el depósito de colágeno, mejora la función endotelial, reduce la inflamación y aumenta la perfusión del miocardio138,139,140. Tras la presentación de dos ensayos controlados y aleatorizados clave, el Randomized Aldactone Evaluation Study (RALES) y el Eplerenone Postacute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS), se añadió el bloqueo de la aldosterona a las guías clínicas para el tratamiento de la insuficiencia cardiaca crónica141,142. Se recomienda la adición de un antagonista de aldosterona en algunos pacientes seleccionados, con síntomas moderadamente graves o graves de insuficiencia cardiaca y reducción de la FEVI o disfunción del VI en la fase inicial tras un infarto de miocardio, cuando puede realizarse una vigilancia cuidadosa de la preservación de la función renal y la concentración de potasio normal143. La efectividad del bloqueo de la aldosterona en la disfunción diastólica y en la insuficiencia cardiaca de leve a moderada no está clara140,144.
Visión general del feocromocitomaLos feocromocitomas son tumores productores de catecolaminas que tienen su origen en las células cromafines de la médula suprarrenal y los ganglios simpáticos (paragangliomas secretores de catecolaminas o feocromocitomas no suprarrenales). La prevalencia estimada del feocromocitoma es de un 0,05-0,12% en la población general y un 0,2-0,6% en los pacientes con hipertensión145. Los pacientes pueden estar asintomáticos si se los diagnostica tras la detección mediante técnicas de imagen suprarrenales o pruebas genéticas. Los pacientes sintomáticos presentan hipertensión (episódica o persistente) y síntomas paroxísticos como mareo, cefalea, rubefacción, diaforesis y palpitaciones. El diagnóstico del feocromocitoma se realiza mediante la confirmación bioquímica de exceso de catecolaminas utilizando las determinaciones urinaria y plasmática de metanefrinas y catecolaminas, seguida de un estudio radiológico para la localización del tumor. El tratamiento del feocromocitoma consiste en la extirpación quirúrgica, con optimización médica preoperatoria para obtener un control adecuado de la presión arterial y una expansión de volumen146,147.
Feocromocitoma y enfermedad cardiovascularRiesgo cardiovascularTienen hipertensión más del 50% de los pacientes con feocromocitoma, y puede ser persistente o paroxística. Se ha observado mayor variabilidad de la presión arterial en el feocromocitoma en comparación con los pacientes con hipertensión esencial, y ello se asocia a mayor incidencia de lesiones de órganos diana148. Se ha descrito resolución de la hipertensión en alrededor del 50% de los pacientes tras el tratamiento quirúrgico satisfactorio del feocromocitoma149.
Se han identificado marcadores de la disfunción endotelial como el aumento del GIM de la carótida en pacientes con feocromocitoma150.
Estos cambios se han atribuido a los efectos del exceso de catecolaminas en el crecimiento y el engrosamiento de la pared vascular. Se ha demostrado que la normalización de los valores de catecolaminas tras la extirpación quirúrgica del feocromocitoma mejora el GIM carotídeo y reduce la fibrosis de la pared de la carótida151.
Estructura y función cardiacasLa acción del exceso de catecolaminas en el feocromocitoma puede conducir a miocardiopatía, cardiopatía isquémica, aturdimiento miocárdico y, excepcionalmente, shock cardiogénico. La incidencia de miocardiopatía en los pacientes con feocromocitoma es de alrededor del 26%; sus manifestaciones principales son miocardiopatía dilatada y miocardiopatía hipertrófica152. La ecocardiografía puede mostrar una dilatación ventricular izquierda con reducción difusa de la contractilidad, dilatación auricular izquierda con aumento de la presión telediastólica, reducción de la fracción de eyección e hipertrofia de tabique. En el contexto de depleción de volumen intravascular y deterioro del llenado diastólico, los pacientes pueden presentar obstrucción al flujo de salida semejante a una miocardiopatía hipertrófica obstructiva. Se observa con frecuencia HVI en la ecocardiografía, y es atribuible en mayor medida a la hipertensión que a los efectos de las catecolaminas150.
Los pacientes con una miocardiopatía asociada a feocromocitoma pueden presentar edema pulmonar o dolor torácico agudo e isquemia/infarto de miocardio. El edema pulmonar se debe a aumento de la permeabilidad capilar pulmonar, aumento de la resistencia vascular periférica, aumento de la presión hidrostática y llenado excesivo o constricción de las venas pulmonares eferentes. La isquemia o el infarto de miocardio pueden ser consecuencia de un vasospasmo coronario, puesto que la acción de las catecolaminas conduce a vasoconstricción, reducción del flujo sanguíneo coronario y aumento de la demanda de oxígeno. Se ha descrito aturdimiento miocárdico tras el vasospasmo inducido por catecolaminas, y se han presentado casos de discinesia apical tipo tako-tsubo causantes de shock cardiogénico agudo153.
Se ha demostrado que la miocardiopatía inducida por catecolaminas mejora tras el tratamiento quirúrgico del feocromocitoma. La reversión de la miocardiopatía depende de la identificación y el tratamiento tempranos. El pronóstico de los pacientes con insuficiencia cardiaca aguda y una lesión miocárdica significativa es muy malo.
RitmoLos signos electrocardiográficos relacionados con el feocromocitoma son desviación del eje a la derecha, mala progresión de la onda R, ondas T invertidas y prolongación del QT. Si hay una lesión permanente del miocardio y se produce una miocardiopatía, puede haber signos de hipertrofia ventricular e isquemia en el electrocardiograma. Pueden observarse arritmias cardiacas en un 20% de los pacientes con feocromocitoma, y pueden consistir en taquicardia sinusal, síndrome del seno enfermo o taquicardia supraventricular y ventricular150,152.
ConclusionesLa disfunción endocrina puede tener repercusiones importantes en el sistema cardiovascular. El restablecimiento de una función endocrina normal conduce con frecuencia a la reversión de las alteraciones cardiovasculares adversas. Las alteraciones cardiacas que se producen a través de efectos hormonales deben tenerse en cuenta al evaluar a pacientes endocrinos y cardiacos.
Conflicto de interesesNinguno.
Autor para correspondencia: 88 East Newton Street, Evans 201, Boston, MA 02118, Estados Unidos. elizabeth.pearce@bmc.org