La principal causa de morbimortalidad en los síndromes de Marfan (SM) y de Loeys-Dietz (SLD) es la dilatación progresiva de la raíz aórtica1,2. El estudio de familiares es crucial tanto para el diagnóstico precoz como para el asesoramiento genético, por lo que deben atenderles equipos multidisciplinares específicos.
Se presenta nuestra experiencia en el tratamiento y el seguimiento de pacientes pediátricos con diagnóstico de SM y SLD. Se siguió en cardiología pediátrica, desde 2005 hasta 2016, a un total de 64 pacientes menores de edad: 52 (81%) con SM clásico, 2 (3%) con SM neonatal y 10 (16%) con SLD. El trabajo coordinado con genetistas y cardiólogos de adultos, así como oftalmólogos, traumatólogos y rehabilitadores, es esencial para una atención integral a estas familias.
Según los criterios de Ghent3, 52 pacientes cumplían criterios de SM clásico. Se estudió a la mitad de ellos (55,8%, 29/52) a raíz de antecedentes familiares conocidos, incluso intraútero. A la otra mitad (44,2%, 23/52), se la diagnosticó por un fenotipo peculiar. De estos, 12 (18,8%) eran casos de novo con estudio genético de ambos progenitores negativo; 10 (15,6%) eran casos índice a partir de los cuales se diagnosticó a algún familiar. De un paciente se descononocían los antecedentes familiares por ser adoptado. En resumen, el 75% tenía afección familiar, datos que concuerdan con la literatura2.
La mayoría de los pacientes tienen confirmación genética FBN1 (80,8%) o está en curso (n = 2). De 7 pacientes no se realizó estudio genético porque cumplían criterios clínicos y familiares.
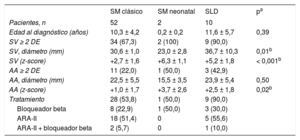
El 67% de los pacientes presentaban dilatación de los senos de Valsalva (SV) (tabla), datos que concuerdan con la literatura que refleja dilatación progresiva en el 50-83% de los pacientes pediátricos2. Las indicaciones de iniciar tratamiento fueron un tamaño aórtico ajustado por z-score4 > +2, excepto para los pacientes menores de 12 años, para quienes se toleraron discretas dilataciones por el bajo riesgo de disección. Tanto los bloqueadores beta como los antagonistas del receptor de la angiotensina II se describieron igualmente eficaces en adultos5 y se indicaron indistintamente a nuestros pacientes (tabla).
Datos ecocardiográficos y tratamiento farmacológico
| SM clásico | SM neonatal | SLD | pa | |
|---|---|---|---|---|
| Pacientes, n | 52 | 2 | 10 | |
| Edad al diagnóstico (años) | 10,3 ± 4,2 | 0,2 ± 0,2 | 11,6 ± 5,7 | 0,39 |
| SV ≥ 2 DE | 34 (67,3) | 2 (100) | 9 (90,0) | |
| SV, diámetro (mm) | 30,6 ± 1,0 | 23,0 ± 2,8 | 36,7 ± 10,3 | 0,01b |
| SV (z-score) | +2,7 ± 1,6 | +6,3 ± 1,1 | +5,2 ± 1,8 | < 0,001b |
| AA ≥ 2 DE | 11 (22,0) | 1 (50,0) | 3 (42,9) | |
| AA, diámetro (mm) | 22,5 ± 5,5 | 15,5 ± 3,5 | 23,9 ± 5,4 | 0,50 |
| AA (z-score) | +1,0 ± 1,7 | +3,7 ± 2,6 | +2,5 ± 1,8 | 0,02b |
| Tratamiento | 28 (53,8) | 1 (50,0) | 9 (90,0) | |
| Bloqueador beta | 8 (22,9) | 1 (50,0) | 3 (30,0) | |
| ARA-II | 18 (51,4) | 0 | 5 (55,6) | |
| ARA-II + bloqueador beta | 2 (5,7) | 0 | 1 (10,0) |
AA: aorta ascendente; ARA–II: antagonista del receptor de la angiotensina II; DE: desviación estándar; SLD: síndrome de Loeys-Dietz; SM: síndrome de Marfan; SV: seno de Valsalva.
Salvo otra indicación, los valores expresan n (%) o media ± DE.
Ningún paciente falleció y 2 precisaron cirugía cardiaca en la edad pediátrica (3,8%). Una adolescente de 13,3 años precisó sustitución de la raíz aórtica según técnica de David (SV, 47 mm; z-score, +6,3) y 1 niño de 6 años precisó una sustitución valvular mitral por insuficiencia mitral grave.
Los 2 casos con SM neonatal se diagnosticaron al nacimiento y tuvieron un curso rápidamente progresivo. Ninguno tenía antecedentes familiares. Uno falleció a los 4,5 meses tras un reemplazo valvular mitral por insuficiencia mitral grave. El otro aún se encuentra en su primer año de vida, con dilatación grave de la raíz aórtica e insuficiencia mitral y tricuspídea moderada-grave en tratamiento médico. El SM neonatal es infrecuente y fenotípicamente muy grave, con dilatación de la raíz aórtica en el 93% y una mortalidad que alcanza el 95% en el primer año de vida2.
Los 10 pacientes con SLD diagnosticados en la edad pediátrica formaban parte de 8 familias. A 2 hermanos se los diagnosticó a raíz de la muerte súbita de su madre por disección aórtica. A los 12,5 años, la hermana menor falleció por disección aórtica (SV, 49 mm; z-score, +7,3) tras rechazar una intervención preventiva. El hermano mayor recibió recambio de la raíz aórtica según técnica de David a los 14 años (SV, 41 mm; z-score, +5,4), pero falleció 5 años más tarde por una hemorragia subaracnoidea. En las otras 7 familias había 8 pacientes afectados; a 3 de ellos se los diagnosticó por tener familiares afectados, 3 eran casos de novo y 2 eran casos índice por los que se diagnosticó a familiares. Todos los pacientes tenían estudio genético positivo (4 TGFBR1, 5 TGFBR2) a excepción de la adolescente que falleció antes de realizarse el estudio (hermano TGFBR1).
La dilatación de los SV y la aorta ascendente de los pacientes con SLD era significativamente más grave que la de los pacientes con SM (tabla). Solo 1 paciente (13 años) tenía tamaño aórtico normal.
Todos los pacientes con SLD que presentaban dilatación (z-score4,6 > +2) recibieron tratamiento con antagonistas del receptor de la angiotensina II (tabla). Precisaron cirugía cardiaca 3 pacientes (30%), a los 12, 14 y 17 años, con recambio de la raíz aórtica; 2 según técnica de David (SV, 42 y 41mm; z-score, +4,7 y +5,4 respectivamente) y 1 según técnica de Bentall (realizado en otro centro). En la edad pediátrica solo 1 paciente falleció (el que rechazó la intervención).
El SLD presenta mayor riesgo de disección aórtica y hemorragia cerebral incluso en la edad pediátrica1. Se recomienda realizar periódicamente pruebas de imagen de todo el árbol arterial, preferiblemente angiorresonancia magnética en niños.
En conclusión, el seguimiento de pacientes con SM y SLD en unidades especializadas en enfermedades cardiacas hereditarias es esencial para una atención integral de estas familias. Hasta el 20% de los pacientes pediátricos permitieron el diagnóstico de un familiar afectado y el 55% de los casos pediátricos se diagnosticaron a partir de un familiar afectado. El diagnóstico y el tratamiento en edades tempranas pueden modificar la evolución natural de la enfermedad, que puede ser especialmente grave y precoz en el SM neonatal y en el SLD.