Las desminopatías son un grupo de enfermedades raras producidas por mutaciones en el gen de la desmina, la gran mayoría autosómicas dominantes, caracterizadas por miopatía esquelética y miocardiopatía (principalmente restrictiva) con arritmias o trastornos en la conducción, debido a que la desmina es el principal componente de los filamentos intermedios en el músculo cardiaco, esquelético y liso, y de las fibras de Purkinje1–3.
El objetivo es analizar el genotipo y fenotipo de pacientes afectos de miocardiopatía asociada a mutaciones en desmina. Las series publicadas incluyen habitualmente a muy pocos pacientes, por lo que cualquier contribución puede ayudar a comprender mejor esta enfermedad.
En 2 centros con 819 familias estudiadas, se analizó a las que, tras el estudio genético guiado por el fenotipo (miocardiopatía restrictiva/dilatada con patrón restrictivo, familias con alta tasa de implante de marcapasos, miopatía esquelética y/o elevación de creatincinasa), presentaban una mutación en desmina. La secuenciación del gen se realizó mediante Sanger o next-generation sequencing. Se consideró mutación patógena a un cambio en la secuencia de aminoácidos respecto a las de referencia cuando cumplía 3 criterios: segregaba con los miembros de la familia afectados, no estaba presente en 200 cromosomas de individuos sanos no relacionados y era un residuo conservado entre especies e isoformas de desmina.
Estudiamos a 20 pacientes de 4 familias, en las que identificamos 3 mutaciones patogénicas: Ile367Phe (2 familias, Sanger), Pro419Ser (Sanger) y Arg415Gln (next-generation sequencing). De estos, 16 presentaban desminopatía (2 portadores obligados fallecidos por miocardiopatía, sin confirmación genética) y 4 eran portadores jóvenes no afectados (tabla). La media de edad al diagnóstico era 35±15 años. Dos familias indicaron a varios familiares portadores de marcapasos y fallecidos de muerte súbita en la tercera o la cuarta década. La clínica más común fue la insuficiencia cardiaca con trastornos de la conducción auriculoventricular. El patrón ecocardiográfico más común fue la miocardiopatía restrictiva, con grosor miocárdico normal o hipertrofia leve (11,4±2,4mm) y función sistólica del ventrículo izquierdo (VI) conservada, salvo 2 pacientes que sufrieron disfunción del VI grave y otro con fenotipo de miocardiopatía arritmogénica del VI (figura). Se realizó cardiorresonancia a 4 pacientes, todos con fibrosis transmural extensa; 7/11 varones y 3/5 mujeres precisaron marcapasos por bloqueo auriculoventricular a edades precoces, incluso 2 varones requirieron resincronización ventricular y 3 desfibriladores automáticos implantables por taquicardia ventricular no sostenida.
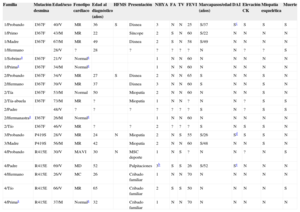
Datos clínicos y genéticos de las familias y los pacientes incluidos en el estudio
| Familia | Mutación desmina | Edad/sexo | Fenotipo cardiaco | Edad al diagnóstico (años) | HFMS | Presentación | NHYA | FA | TV | FEVI | Marcapasos/edad (años) | DAI | Elevación CK | Miopatía esquelética | Muerte |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1/Probando | I367F | 40/V | MR | 36 | S | Disnea | 3 | N | N | 25 | S/37 | Sc | S | S | S |
| 1/Primo | I367F | 43/M | MR | 22 | Síncope | 2 | S | N | 60 | S/22 | N | N | N | N | |
| 1/Madre | I367F | 67/M | MR | 49 | Disnea | 2 | S | N | 58 | S/49 | N | N | N | N | |
| 1/Hermano | 28/V | ? | 28 | ? | ? | ? | ? | ? | N | N | ? | ? | S | ||
| 1/Sobrinoa | I367F | 21/V | Normala | 1 | N | N | 60 | N | N | N | N | N | |||
| 1/Primaa | I367F | 34/M | Normala | 1 | N | N | 60 | N | N | N | N | N | |||
| 2/Probando | I367F | 34/V | MR | 27 | S | Disnea | 2 | N | N | 65 | S | N | N | S | N |
| 2/Hermano | I367F | 39/V | MR | 37 | Disnea | 3 | N | N | 60 | S | N | N | S | N | |
| 2/Tía | I367F | 53/M | Normal | 50 | Miopatía | 2 | N | N | 60 | N | N | N | S | N | |
| 2/Tía-abuela | I367F | 73/M | MR | ? | Miopatía | 1 | N | N | ? | N | N | ? | S | S | |
| 2/Padre | 48/V | ? | ? | ? | ? | ? | ? | ? | S | N | ? | S | S | ||
| 2/Hermanastraa | I367F | 26/M | Normala | 1 | N | N | 60 | N | N | N | N | N | |||
| 2/Tío | I367F | 46/V | MR | ? | ? | 2 | ? | ? | ? | S | N | S | S | N | |
| 3/Probando | P419S | 28/V | MR | 24 | N | Miopatía | 2 | N | S | 55 | S/26 | Sd | S | S | N |
| 3/Madre | P419S | 56/M | MR | 42 | Miopatía | 2 | N | N | 60 | S/48 | N | N | S | N | |
| 4/Probando | R415E | 30/V | MAVI | 30 | N | MSC deporte | 1 | N | S | ? | N | N | ? | N | S |
| 4/Padre | R415E | 69/V | MD | 52 | Palpitaciones | 3b | S | S | 26 | S/52 | Se | N | N | N | |
| 4/Hermano | R415E | 26/V | MC | 26 | Cribado familiar | 1 | N | N | 70 | N | N | N | N | N | |
| 4/Tío | R415E | 66/V | MR | 65 | Cribado familiar | 2 | S | S | 50 | N | N | N | N | S | |
| 4/Primaa | R415E | 37/M | Normala | 32 | Cribado familiar | 1 | N | N | 70 | N | N | N | N | N |
CK: creatincinasa; DAI: desfibrilador automático implantable; FA: fibrilación auricular; FEVI: fracción de eyección del ventrículo izquierdo; HFMS: historia familiar de muerte súbita; I367F: Ile367Phe; M: mujer; MAVI: miocardiopatía arritmogénica de ventrículo izquierdo; MC: miocardiopatía no especificada; MD: miocardiopatía dilatada; MR: miocardiopatía restrictiva; MSC: muerte súbita cardiaca; NHYA: clasificación de la disnea de la New York Heart Association; N: no; P419S: Pro419Ser; R415E: Arg415Glu; S: sí; TV: taquicardia ventricular; TVNS, taquicardia ventricular no sostenida; V: varón.
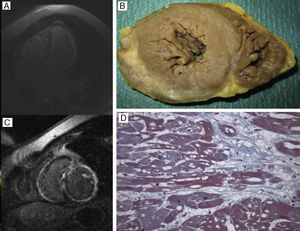
, que muestra dilatación biauricular y biventricular y fibrosis extensa transmural en pared libre del ventrículo izquierdo, septo y ventrículo derecho. B y D: necropsia del probando de la familia 4 (mutación DES Arg415Glu), que muestra grosores y diámetros normales, hipertrofia fibrilar y fibrosis subepicárdica grave, vacuolización citoplásmica y pérdida miofibrilar, todo ello compatible con miocardiopatía arritmogénica ventricular izquierda.")
A y C: cardiorresonancia del probando de la familia 1 (mutación DES Ile367Phe), que muestra dilatación biauricular y biventricular y fibrosis extensa transmural en pared libre del ventrículo izquierdo, septo y ventrículo derecho. B y D: necropsia del probando de la familia 4 (mutación DES Arg415Glu), que muestra grosores y diámetros normales, hipertrofia fibrilar y fibrosis subepicárdica grave, vacuolización citoplásmica y pérdida miofibrilar, todo ello compatible con miocardiopatía arritmogénica ventricular izquierda.
El grado de miopatía esquelética fue variable, principalmente debilidad y atrofia muscular distal y progresiva de extremidades inferiores. Hubo una elevada penetrancia a partir de la tercera década, con expresión más grave en varones.
Seis pacientes fallecieron en el seguimiento: 1 por muerte súbita (varón de 30 años, miocardiopatía arritmogénica VI), 2 por insuficiencia cardiaca (varón de 40 años con disfunción del VI grave y 1 varón de 66 con miocardiopatía restrictiva) y 3 por causas desconocidas (1 portador de marcapasos).
Se conocen pocos datos de estas 3 mutaciones, ya que solo están descritas por la literatura en 4 familias: 1 familia Ile367Phe1,2, 3 Pro419Ser1,2,4 y ninguna Arg415Glu.
La mutación Ile367Phe se ha descrito previamente como patogénica2. En nuestras 2 familias comprobamos la segregación de la mutación con la enfermedad. El fenotipo es común al ya descrito: miocardiopatía restrictiva, bloqueo auriculoventricular y miopatía esquelética.
La mutación Pro419Ser también se ha descrito y presenta un fenotipo más variable: afección neurológica marcada con predominio de debilidad muscular distal y voz nasal, miocardiopatía restrictiva y bloqueo auriculoventricular, y en otra familia miocardiopatía arritmogénica del ventrículo derecho2,4. Esta mutación se localiza en la cola del gen, comúnmente aparece la miocardiopatía sola o seguida de afección esquelética. Por el contrario, la Ile367Phe está situada en un hot spot y reviste mayor gravedad2.
La mutación Arg415Glu en el exón 6 no se ha descrito antes y afecta al splicing. En nuestra familia se demuestra la cosegregación. El probando falleció de muerte súbita, con hallazgos histológicos de miocardiopatía arritmogénica del VI, y es el primer caso publicado de esta asociación. Esta mutación produjo en la mayoría de nuestros pacientes miocardiopatía grave pero sin miopatía esquelética.
Destaca que estos pacientes llegaron a la consulta con otros diagnósticos, como cardiopatía hipertensiva en fase dilatada o miocardiopatía hipertrófica. El cuarto probando fue diagnosticado tras muerte súbita. El estudio especializado permitió sospechar su origen, confirmado mediante genética. El posterior estudio familiar ha permitido diagnosticar a 14 portadores, 10 con miocardiopatía restrictiva y/o miopatía esquelética y 4 jóvenes portadores no afectados. Asimismo ha permitido excluir del seguimiento a 16 familiares.
Se concluye que las desminopatías se presentan comúnmente como miocardiopatía restrictiva con insuficiencia cardiaca en la tercera o la cuarta década, bloqueo auriculoventricular avanzado precoz que obliga a implante de marcapasos y en algunos casos, además, desfibrilador automático implantable por taquicardia ventricular no sostenida.
FINANCIACIÓNRed Investigación Cardiovascular (RIC) del Instituto de Salud Carlos III (PI14/01477, RD12/0042/0029, RD12/0042/0069), FEDER y CIBEROBN (CB12/03/30038), Madrid, España.