En 4 miembros de una familia española se identificó una mutación en los canales cardiacos Nav1.5 (p.R1644H) descrita ya y relacionada con el síndrome de QT largo con anterioridad. Sin embargo, solo 1 de los portadores presentaba el intervalo QT prolongado. En los otros 3 individuos se identificó una nueva mutación con cambio de sentido en los canales cardiacos Cav1.2 (p.S1961N). En este trabajo se analizaron las características funcionales de los canales p.S1961N Cav1.2 para averiguar si dicha mutación regula la expresividad del síndrome de QT largo en esta familia.
MétodosLa corriente de calcio tipo L (ICaL) se registró mediante la técnica de patch-clamp en células de ovario de hámster chino transfectadas transitoriamente con los canales cardiacos humanos en su forma nativa o mutada.
ResultadosLa expresión de canales p.S1961N disminuye significativamente la densidad de la ICaL. Al sustituir el ion calcio por bario para suprimir la inactivación dependiente del calcio de los canales Cav1.2, se demostró que la mutación acelera significativamente la inactivación dependiente del voltaje de los canales Cav1.2 y disminuye la constante de tiempo de inactivación. Como consecuencia, la carga total que atraviesa los canales p.S1961N Cav1.2 disminuye significativamente. Los efectos que las mutaciones p.S1961N Cav1.2 y p.R1644H Nav1.5, por separado o en combinación, producen sobre las características de los potenciales de acción (PA) se simularon mediante un modelo matemático de PA ventriculares humanos. Los resultados demuestran que la mutación p.S1961N Cav1.2 abrevia la duración del PA y suprime la prolongación inducida por la mutación p.R1644H de los canales Nav1.5.
ConclusionesLa mutación p.S1961N en los canales Cav1.2 disminuye la ICaL, un efecto que podría abreviar la duración de los PA ventriculares humanos. La presencia de esta mutación que disminuye la función de los canales Cav1.2 compensa funcionalmente los efectos producidos por la mutación de los canales Nav1.5 que aumenta su función y prolonga la duración de los PA.
Palabras clave
El síndrome de QT largo (SQTL) es un síndrome arritmógeno primario y hereditario que se caracteriza por un intervalo QT prolongado en el electrocardiograma (ECG), causado por un retraso en la repolarización ventricular y asociado con síncope, convulsiones, taquicardia ventricular (principalmente torsades de pointes) y alto riesgo de muerte súbita cardiaca1,2. La duración del potencial de acción (DPA) ventricular es el resultado de un equilibrio entre las corrientes despolarizantes (Na y Ca) y repolarizantes (K). En consecuencia, tanto un aumento en las corrientes despolarizantes como una disminución en las corrientes repolarizantes podrían originar los distintos tipos de SQTL1,2.
El SQTL de tipo 3 se debe a mutaciones de ganancia de función en el gen SCN5A, que codifica la subunidad α de los canales de sodio (Nav1.5) encargados de la corriente cardiaca de sodio (INa)3. Estas mutaciones suelen modificar la cinética de los canales Nav1.5 por medio de diversos mecanismos biofísicos que conducen en última instancia a un aumento del componente lento o sostenido de la INa (INaL) que genera una entrada de Na persistente durante la fase de meseta del PA ventricular que prolonga la DPA3.
Los genes CACNA1C, CACNB2 y CACNA2D1 codifican la subunidad α formadora del poro (Cav1.2) y las subunidades auxiliares β2 y α2δ, respectivamente, que constituyen el canal de Ca cardiaco que genera la corriente de Ca de tipo L (ICaL)4. Las mutaciones de pérdida de función en los genes CACNA1C y CACNB2 acortan la DPA y se han asociado con el síndrome de Brugada y el síndrome de QT corto5,6. Por el contrario, las mutaciones de ganancia de función en el gen CACNA1C producen la prolongación de la DPA ventricular y SQTL de tipo 87.
En 4 miembros de una familia española, en la que solo 1 de los portadores presentaba un fenotipo de SQTL de tipo 3, se identificó una mutación de ganancia de función en el gen SCN5A que codifica los canales p.R1644H Nav1.5 ya descrita previamente8. Los análisis genéticos de los otros 3 miembros que presentaban valores QT corregidos (QTc) normales demostraron que, además, eran portadores de una nueva mutación con cambio de sentido en el gen CACNA1C, que codifica los canales p.S1961N Cav1.2. En consecuencia, estos miembros de la familia son portadores de 2 variantes en distintos genes (heterocigosis digénica). Así pues, el objetivo de este estudio es caracterizar las propiedades electrofisiológicas de los canales p.S1961N Cav1.2 para dilucidar si la presencia de esta variante podría compensar la prolongación de la DPA producida por la mutación p.R1644H Nav1.5 y con ello el SQTL.
MÉTODOSAnálisis genéticoSe realizó la evaluación clínica, incluido un ECG de 12 derivaciones, de los miembros de la familia en el Hospital Universitario Virgen de las Nieves, Granada. El estudio fue aprobado por el comité de ética local y se realizó de acuerdo con los principios indicados en la Declaración de Helsinki. Cada participante dio su consentimiento informado por escrito9–11.
Toda la secuencia codificante y las regiones intrónicas complementarias de los genes KCNQ1, KCNH2, SCN5A, KCNJ2, KCNJ8, CACNA1C, AKAP9, KCNE1 y KCNE2 se secuenciaron utilizando una plataforma de secuenciación de segunda generación Hiseq 1500 (Illumina). Las variantes potencialmente patógenas se confirmaron utilizando la secuenciación de Sanger9–11. La patogenicidad de las variantes se predijo según las recomendaciones actuales del American College of Medical Genetics and Genomics y la Association for Molecular Pathology (véanse detalles en el apartado «Análisis genético» del material suplementario)12. Se hallaron otras variantes exónicas no sinónimas, no predichas como patógenas, que se describen en la tabla del material suplementario.
Mutagénesis y transfección celularLa sustitución de p.S1961N en Cav1.2 (NP_000710.5) se llevó a cabo utilizando el kit de mutagénesis dirigida QuikChange (Stratagene, Estados Unidos) y se confirmó mediante secuenciación directa de ácido desoxirribonucleico (ADN)5,9,11,13. Se transfectaron transitoriamente células de ovario de hámster chino con el ADNc que codifica los canales Cav1.2 nativos (WT) o mutados y las subunidades α2δ y β (en proporción 1:1,7:4).
Técnica de patch-clampLas corrientes se registraron utilizando la configuración «célula entera» de la técnica de «fijación de membrana» (patch-clamp) siguiendo los métodos descritos anteriormente9,13. La resistencia de acceso, la capacitancia celular y la amplitud máxima de la ICaL generadas por los canales Cav1.2 WT fueron de 1,8 ± 0,4 MΩ, 16,2 ± 1,2 pF y –521 ± 60 pA (n = 25) respectivamente. Por lo tanto, no se esperaban errores de voltaje significativos (< 5 mV) debidos a la resistencia en serie con las micropipetas utilizadas.
Modelo matemático del potencial de acción ventricularPara simular las condiciones del PA ventricular humano, se empleó el modelo de O’Hara-Rudy validado anteriormente y utilizado con finalidades parecidas14. El modelo se ejecutó a distintas frecuencias (0,1-3 Hz) en condiciones de control (WT) o incorporando los cambios en la INaL y la ICaL producidos por las mutaciones p.R1644H Nav1.5 y p.S1961N Cav1.2 respectivamente.
Análisis estadísticoLos resultados se expresan como media ± error estándar de la media. Cuando fue apropiado se recurrió a la prueba de la t de Student apareada o desapareada o ANOVA unifactorial, seguido de la prueba de Newman-Keuls para evaluar la significación estadística. Para incluir las evaluaciones sucesivas de las muestras, los datos se analizaron con modelos multinivel de efectos mixtos. Un valor de p < 0,05 se consideró estadísticamente significativo. En el material suplementario está disponible un apartado de «Métodos» ampliado.
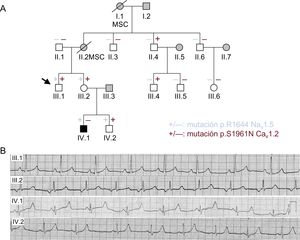
RESULTADOSDescripción de los casos y análisis genéticoEl probando (III.1) (figura 1A, flecha) sufrió un episodio sincopal cuando tenía 28 años, después de haber tomado una única dosis de ciprofloxacino, un fármaco que prolonga el QT15. Esto indicaba que podría tener predisposición genética a la prolongación del QT inducida por fármacos. Por el contrario, la evaluación clínica reveló que presentaba un ECG normal con un QTc de Bazett de 420ms (figura 1B). No obstante, tanto su madre (II.2) como su abuela (I.1), que no habían sufrido nunca episodios sincopales, fallecieron de manera súbita estando en reposo a la edad de 49 años (no se dispone de datos clínicos o genéticos). En consecuencia, los antecedentes familiares de muerte súbita cardiaca propiciaron la evaluación de los familiares del probando. La hermana del probando (III.2) mostró un ritmo auricular inferior a 60 lpm, duraciones PR y QRS normales, y una onda T ligeramente prolongada (figuras 1A y B). No obstante, el QT disminuyó correctamente durante el ejercicio y el QTc fue también normal (430 ms). Uno de los sobrinos del probando (IV.1), un chico de 13 años, mostró un ECG con una onda T tardía tras un segmento ST plano y largo, lo que resultó en un QTc notablemente prolongado (558 ms) (figura 1B). Por el contrario, el individuo IV.2 mostró un QTc de 420ms con una onda T normal (figura 1B). El resto de los familiares estudiados (figura 1A, símbolos blancos) han permanecido y permanecen actualmente asintomáticos, con ECG completamente normales.
![A: árbol genealógico de la familia (mujeres [○] y varones [□]). La flecha indica el probando. Las líneas en diagonal indican los pacientes fallecidos. + y – indican individuos con y sin las variantes p.R1644H Nav1.5 (azul) y p.S1961N Cav1.2 (rojo) respectivamente. Los símbolos en negro indican individuos afectados por el síndrome de QT largo, mientras que los símbolos en gris representan a individuos que no se han sometido a pruebas genéticas. Los símbolos en blanco representan a individuos estudiados sin ningún fenotipo clínico. B: electrocardiograma de los pacientes III.1, III.2, IV.1 y IV.2. MSC: muerte súbita cardiaca. Esta figura se muestra a todo color solo en la versión electrónica del artículo.](https://static.elsevier.es/multimedia/03008932/0000007200000004/v1_201903190611/S0300893218301180/v1_201903190611/es/main.assets/gr1.jpeg?xkr=eyJpdiI6IkM3V21vbWpYTkhMZDNJdmpBRE5TaFE9PSIsInZhbHVlIjoiZ0duNWM4VjAzMVlBZlE1c2pzRjlnMStHUFo2QSs0UEk0MDh4K0kyQU41YXRDUEpNaXdlYU8zZ3JUZFk5ZEhMNWdaTEtHdGg4UFpJN2lVVERHTlVKSC82TEZHUjhXM3BzMGF6TGh6NWtDdHBNa281aXJISVBOTTNSbjVoYzAyWTVNd0pmdlpzejdiOTBhV3UvcnBtM3ptaXVZZHgwZzlqNDgwSXZKR2l5L0N4WHgzV3VRNUxxQlNrb1BUdmNEY05QcGRoRGdLaE9vT1EzR3FLaHlnb3RaMTI3MEx5MXNQSFBnbTlhb2hzVy9uR0lPQlMwWlVxdFAvYXVVb0pmR3RKMjd2Z0s1dXZOQm5wN1dTczIwWmdMby8rSDE0c3JGcDBDUTd6aFhLZHRFbXM9IiwibWFjIjoiZDc0NTViMTg1MmVlMTFkZmFjYzVmMzJlM2M4MDRjODBkOWI3OGY0YzY4YjRlN2MyOGQyNjljNzRlNjhhMzk0ZSIsInRhZyI6IiJ9 "A: árbol genealógico de la familia (mujeres [○] y varones [□]). La flecha indica el probando. Las líneas en diagonal indican los pacientes fallecidos. + y – indican individuos con y sin las variantes p.R1644H Nav1.5 (azul) y p.S1961N Cav1.2 (rojo) respectivamente. Los símbolos en negro indican individuos afectados por el síndrome de QT largo, mientras que los símbolos en gris representan a individuos que no se han sometido a pruebas genéticas. Los símbolos en blanco representan a individuos estudiados sin ningún fenotipo clínico. B: electrocardiograma de los pacientes III.1, III.2, IV.1 y IV.2. MSC: muerte súbita cardiaca. Esta figura se muestra a todo color solo en la versión electrónica del artículo.")
A: árbol genealógico de la familia (mujeres [○] y varones [□]). La flecha indica el probando. Las líneas en diagonal indican los pacientes fallecidos. + y – indican individuos con y sin las variantes p.R1644H Nav1.5 (azul) y p.S1961N Cav1.2 (rojo) respectivamente. Los símbolos en negro indican individuos afectados por el síndrome de QT largo, mientras que los símbolos en gris representan a individuos que no se han sometido a pruebas genéticas. Los símbolos en blanco representan a individuos estudiados sin ningún fenotipo clínico. B: electrocardiograma de los pacientes III.1, III.2, IV.1 y IV.2. MSC: muerte súbita cardiaca. Esta figura se muestra a todo color solo en la versión electrónica del artículo.
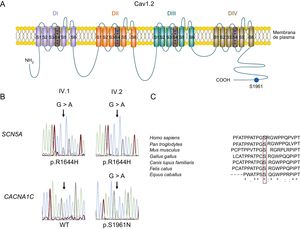
Teniendo en cuenta los datos clínicos, se ofreció un análisis genético mediante secuenciación masiva de segunda generación (véase «Métodos») tanto al probando, quien en un inicio lo rechazó, como a su hermana, que aceptó. La genotipificación de la hermana del probando permitió identificar 2 mutaciones (heterocigosis digénica). Estas mutaciones se secuenciaron después mediante el método de Sanger en el probando (que finalmente aceptó) y los demás familiares estudiados. La primera mutación, predicha como patógena (véase el apartado «Análisis genético» del material suplementario), apareció en el gen SCN5A (NC_000003.11:g.38592932C>T según GRCh37) y produce la sustitución de arginina por histidina en el residuo 1644 (p.R1644H) de los canales Nav1.5. Esta mutación ya se había descrito con anterioridad en varias familias con SQTL de tipo 3 y nunca se ha relacionado con ningún otro fenotipo arrítmico, incluido el síndrome de Brugada8,16–18. Los estudios funcionales revelaron que la mutación p.R1644H produce un aumento notable de la INaL (de ganancia de función), lo que explica la prolongación de la DPA y del QT8,16. La mutación p.R1644H Nav1.5 también estaba presente en el probando y sus sobrinos, pero en ningún otro de los familiares estudiados (figura 1A). En la hermana del probando, en este y en 1 de sus sobrinos (figura 1A) también se identificó una mutación en el gen CACNA1C (NC_000012.11:g.2797710G>A) (figura 2B) que produce la sustitución de la serina en la posición 1961 por asparagina (p.S1961N) en el canal Cav1.2 (figura 2A). El residuo de serina en la posición equivalente está muy conservado en las distintas especies (figura 2C) y la mutación no se ha anotado con anterioridad en ninguna base de datos pública. Cabe destacar que los individuos II.4 y III.4 también eran portadores de la mutación p.S1961N Cav1.2, pero ambos presentaban un ECG normal y no habían sufrido nunca un episodio arrítmico.
 del gen SCN5A, que produce la mutación con cambio de sentido p.R1644H en ambos individuos (parte superior), y una secuencia WT (izquierda) y la mutación heterocigótica (NC_000012.11:g.2797710G>A) (derecha) del gen CACNA1C, que da como resultado la mutación con cambio de sentido p.S1961N (parte inferior). C: alineación de secuencias de las regiones que rodean el residuo S1961 de los canales Cav1.2 en varias especies; el recuadro destaca la conservación de este residuo. «*» indica residuos idénticos; «:» y «.» indican la conservación entre los grupos de propiedades muy y algo similares respectivamente. ADN: ácido desoxirribonucleico; WT: forma nativa del canal.")
A: esquema del canal de calcio cardiaco de tipo L. B: cromatogramas de secuencias de ADN de los pacientes IV.1 y IV.2 que representan la variación heterocigótica (NC_000003.11:g.38592932C>T) del gen SCN5A, que produce la mutación con cambio de sentido p.R1644H en ambos individuos (parte superior), y una secuencia WT (izquierda) y la mutación heterocigótica (NC_000012.11:g.2797710G>A) (derecha) del gen CACNA1C, que da como resultado la mutación con cambio de sentido p.S1961N (parte inferior). C: alineación de secuencias de las regiones que rodean el residuo S1961 de los canales Cav1.2 en varias especies; el recuadro destaca la conservación de este residuo. «*» indica residuos idénticos; «:» y «.» indican la conservación entre los grupos de propiedades muy y algo similares respectivamente. ADN: ácido desoxirribonucleico; WT: forma nativa del canal.
En consecuencia, los miembros de la familia que mostraron heterocigosis digénica no presentaban un fenotipo de SQTL de tipo 3, mientras que sí lo tenía IV.1, que es el único portador de solo la mutación en el gen SCN5A. Así pues, se decidió caracterizar funcionalmente la mutación p.S1961N Cav1.2 para dilucidar si la presencia de esta variante puede anular la prolongación de la DPA y compensar el SQTL producido por la mutación p.R1644H Nav1.5.
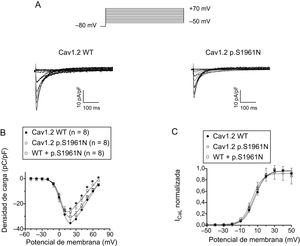
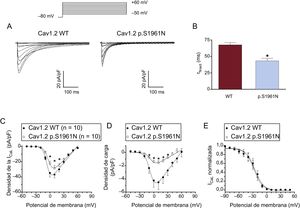
Caracterización funcional de los canales p.S1961N Cav1.2La figura 3A muestra los trazos de la ICaL registrados a potenciales entre –50 y +70mV en células de ovario de hámster chino que expresan canales Cav1.2 cardiacos humanos nativos o mutantes junto con las subunidades α2δ y β. La densidad máxima de las corrientes generadas por los canales Cav1.2 mutantes (–26,4 ± 1,4 pA/pF; p < 0,05) fue aproximadamente un 25% menor que la generada por los canales Cav1.2 nativos (–35,5 ± 3,0 pA/pF) (figuras 3A y B). En otro grupo de experimentos, se transfectaron células con el ADNc que codifica los canales Cav1.2 nativos y mutantes (en proporción 0,5:0,5) considerando que los portadores de la mutación lo son en heterocigosis. En estas condiciones, la densidad de corriente máxima no fue estadísticamente distinta de la generada por los canales nativos y mutantes por separado (figura 3B; p > 0,05). La figura 3C muestra la dependencia de voltaje de la activación de los canales Cav1.2 WT, p.S1961N y WT+p.S1961N obtenida representando la conductancia normalizada (estimada por la ecuación n.o 1; véase el apartado «Patch-clamping» del material suplementario) en función del potencial de membrana. El ajuste de una función de Boltzmann permitió obtener los valores de punto medio (Vh) y pendiente (k) de las curvas, que casi se superponen (figura 3C). En consecuencia, los valores de Vh y de k de las 3 curvas no fueron significativamente distintos (tabla). El ajuste de la ecuación n.o 2 (véase el apartado «Patch-clamping» del material suplementario) a los datos de densidad de corriente en función del voltaje de cada experimento permitió calcular el potencial de inversión (Erev), que resultó ser de 74,9 ± 2,7mV para los canales nativos (WT). Los canales mutantes, ya fuera solos o en combinación con canales nativos (WT), presentaban el mismo valor de Erev (p > 0,05) (tabla).
 y de conductancia (C) en función del voltaje para la ICaL generada por los canales Cav1.2 WT, p.S1961N o WT+p.S1961N; cada punto representa la media ± error estándar de la media de (n) experimentos. En B, *p < 0,05 frente a Cav1.2 WT. ICaL: corriente de Ca de tipo L; WT: forma nativa del canal.")
A: corrientes generadas por los canales Cav1.2 WT o p.S1961N. B y C: curvas de densidad (B) y de conductancia (C) en función del voltaje para la ICaL generada por los canales Cav1.2 WT, p.S1961N o WT+p.S1961N; cada punto representa la media ± error estándar de la media de (n) experimentos. En B, *p < 0,05 frente a Cav1.2 WT. ICaL: corriente de Ca de tipo L; WT: forma nativa del canal.
Propiedades biofísicas de los canales Cav1.2 nativos y mutantes
| Activación | Inactivación | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Constructo Cav1.2 (ICaL) | Carga a +20mV (pC/pF) | Erev(mV) | Vh(mV) | k | τ (ms) | Vh(mV) | k | τrápida(ms) | τlenta(ms) |
| WT (n = 8) | –1,7 ± 0,2 | 74,9 ± 2,7 | 7,3 ± 1,4 | 6,4 ± 0,3 | 1,2 ± 0,03 | –19,1 ± 1,2 | 7,0 ± 0,9 | 22,4 ± 0,3 | 89,4 ± 5,1 |
| p.S1961N (n = 8) | –1,0 ± 0,1a | 73,2 ± 1,3 | 5,5 ± 1,3 | 6,8 ± 0,2 | 1.1 ± 0,1 | –20,6 ± 2,2 | 7,8 ± 0,5 | 14,7 ± 1,1a | 78,3 ± 6,0 |
| WT+p.S1961N (n = 9) | –1,4 ± 0,1a,b | 76,1 ± 0,9 | 6,1 ± 1,6 | 6,9 ± 0,2 | 1.2 ± 0,1 | –21,1 ± 2,3 | 7,9 ± 0,6 | 18,1 ± 1,0a,b | 90,3 ± 3,9 |
| Constructo Cav1.2 (IBa) | Carga a +10mV (pC/pF) | Erev(mV) | Vh(mV) | k | τ (ms) | Vh(mV) | k | τ (ms) | |
|---|---|---|---|---|---|---|---|---|---|
| WT (n = 13) | –4,5 ± 1,1 | 55,7 ± 0,9 | –3,4 ± 1,3 | 4,9 ± 0,4 | 1,8 ± 0,1 | –27,9 ± 4,4 | 8,7 ± 1,6 | 67,5 ± 3,3 | |
| p.S1961N (n = 10) | –1,6 ± 0,3a | 54,8 ± 1,7 | –0,8 ± 1,3 | 5,1 ± 0,3 | 1,5 ± 0,2 | –30,0 ± 4,6 | 7,9 ± 1,0 | 43,1 ± 3,9a |
Erev: potencial de inversión; IBa: corriente generada utilizando Ba como portador de carga; ICaL: corriente de Ca de tipo L; τ: constantes de tiempo de activación (ICaL)/inactivación (IBa) obtenidas ajustando una función monoexponencial a los trazos de corriente registrados a +20 mV; τrápida y τlenta: constantes de tiempo rápida y lenta de la inactivación (ICaL) obtenidas tras ajustar una función biexponencial a los trazos de corriente registrados a +20 mV; Vh y k: valores del punto medio y la pendiente de las curvas de inactivación y activación; WT: forma nativa de los canales.
Los valores expresan la media ± error estándar de la media de (n) experimentos en cada grupo.
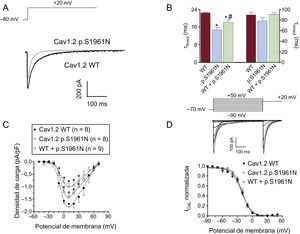
La figura 4A muestra trazos superpuestos de ICaL obtenidos al aplicar pulsos de 500ms desde –80 a +20mV en células que expresan canales Cav1.2 nativos o mutantes (las corrientes generadas por los canales WT+p.S1961N se omitieron para mayor claridad). La cinética de activación de los canales Cav1.2 no fue modificada por la mutación p.S1961N (tabla; p > 0,05). Sin embargo, la mutación aceleró significativamente el curso temporal de la inactivación de la corriente (figura 4A), disminuyendo la constante de tiempo de inactivación rápida (τ rápida) calculada por el ajuste de una función biexponencial a la caída de la corriente (figura 4B y tabla). Curiosamente, la τ rápida de la corriente generada por los canales WT+p.S1961N fue intermedia (figura 4B y tabla). Por lo tanto, para cuantificar los efectos producidos por la mutación p.S1961N en el flujo total de Ca, se midió la densidad de carga generada por los canales Cav1.2 WT, p.S1961N y WT+p.S1961N a cada potencial de membrana (figura 4C). La densidad, por su parte, se calculó normalizando la carga que atraviesa la membrana, estimada a partir de la integral de los trazos de corriente, con respecto a la capacitancia celular (véase el apartado «Métodos» del material suplementario). La mutación p.S1961N en los canales Cav1.2 disminuye de manera significativa la carga que atraviesa la membrana por los efectos combinados de la mutación en la densidad de corriente máxima, y en la cinética de la inactivación, generan una carga (figura 4C).

A: trazos de corriente superpuestos generados a +20mV por los canales Cav1.2 WT o p.S1961N. B: constantes de tiempo rápida y lenta de la inactivación obtenida a +20mV en células que expresan canales Cav1.2 WT, p.S1961N o WT+p.S1961N. C: carga estimada como la integral de los trazos de corriente registrados en función del potencial de membrana en los 3 grupos. D: arriba, corrientes generadas por los canales Cav1.2 WT utilizando el protocolo descrito; abajo, curvas de inactivación para los canales Cav1.2 WT, p.S1961N o WT+p.S1961N; cada línea/punto representa la media ± error estándar de la media de 8 o más experimentos. En B y C, *p < 0,05 frente a Cav1.2 WT. WT: forma nativa del canal.
La figura 4D (parte superior) muestra los trazos de la ICaL generados por los canales Cav1.2 nativos junto con el protocolo utilizado para cuantificar la dependencia de voltaje de la inactivación y describir las curvas de inactivación («Métodos» del material suplementario) (figura 4D, abajo). Ni el Vh ni la k de las curvas de inactivación se modificaron por la mutación p.S1961N sola o combinada con los canales WT (figura 4D y tabla).
La mutación p.S1961N aceleró la inactivación de los Cav1.2 dependientes de voltajeLa inactivación de los canales Cav1.2 consiste en 2 mecanismos independientes, la inactivación dependiente del Ca (IDC) y la inactivación dependiente del voltaje (IDV). Se probaron los efectos de la mutación p.S1961N en la IDV utilizando Ba como transportador de carga, ya que las corrientes de Ba (IBa) no muestran IDC19. La figura 5A muestra los trazados de la IBa generados en 2 células de ovario de hámster chino transfectados con canales Cav1.2 nativos y mutantes respectivamente. El curso temporal de la inactivación de la IBa, definida por una función monoexponencial, se aceleró notablemente por la mutación (figura 5B y tabla). Además, la mutación disminuyó significativamente tanto la IBa máxima como la densidad de la IBa, y la disminución de la carga fue mayor que la de la corriente máxima (figuras 5C y D). Por último, la dependencia del voltaje de la inactivación de la IBa no se modificó por la mutación (figura 5E y tabla). Todos estos resultados indican claramente que la mutación p.S1961N acelera la IDV de los canales Cav1.2.
, curvas obtenidas al representar la densidad de corriente (C) o la densidad de carga (D) en función del voltaje y curvas de inactivación (E) para la IBa generada por lo canales WT o p.S1961N; cada punto representa la media ± error estándar de la media de 10 experimentos. En B y C, *p < 0,05 frente a Cav1.2 WT. WT: forma nativa del canal.")
A: trazos de IBa generados por los canales Cav1.2 WT o p.S1961N. B–E: constantes de tiempo de inactivación obtenidas a +10mV (B), curvas obtenidas al representar la densidad de corriente (C) o la densidad de carga (D) en función del voltaje y curvas de inactivación (E) para la IBa generada por lo canales WT o p.S1961N; cada punto representa la media ± error estándar de la media de 10 experimentos. En B y C, *p < 0,05 frente a Cav1.2 WT. WT: forma nativa del canal.
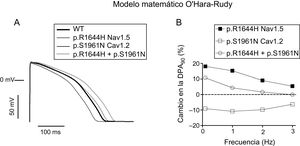
Los efectos de las mutaciones p.S1961N Cav1.2 y p.R1644H Nav1.5, solas o combinadas, en las características del PA se simularon utilizando un modelo matemático ya validado de PA ventriculares endocárdicos humanos (figura 6)14. El modelo se ejecutó en condiciones de control (WT) o introduciendo los cambios cinéticos y en la conductancia producidos por la mutación p.S1961N Cav1.2. Para simular los efectos de la mutación p.R1644H, se aumentó la conductancia de la INaL ≈3 veces, considerando el efecto producido por la mutación descrito con anterioridad8. La figura 6A muestra los trazos de los PA obtenidos al correr el modelo considerando una frecuencia basal de 1Hz. La mutación p.R1644H Nav1.5 prolongó la DPA20, la DPA50 y la DPA90 el 17,4, el 17,1 y el 15,2% respectivamente. Dada la reducción en la ICaL, p.S1961N Cav1.2 acortó la DPA20, la DPA50 y la DPA90 el 2,7, el 14,3 y el 10,8% (figura 6A). Curiosamente, los trazos de los PA correspondientes a p.R1644H+p.S1961N y WT eran prácticamente indistinguibles, y solo se observó una ligera prolongación de la DPA90 (4,3%). La figura 6B representa el cambio en la DPA90 (prolongación o acortamiento) inducido por cada mutación en células estimuladas a distintas frecuencias (0,1-3 Hz). La prolongación inducida por p.R1644H Nav1.5 fue máxima a bajas frecuencias y disminuyó progresivamente a frecuencias más altas. El acortamiento inducido por p.S1961N Cav1.2 fue similar a frecuencias entre 0,1 y 2Hz (≈10%) y disminuyó ligeramente a 3,0Hz. En presencia de p.S1961N, la prolongación inducida por p.R1644H se redujo hasta desaparecer por completo a las frecuencias más altas (2 y 3 Hz).
 o en presencia de las mutaciones p.R1644H Nav1.5 y p.S1961N Cav1.2 por separado o combinadas. B: porcentaje de la prolongación (valores positivos) o el acortamiento (valores negativos) de la DPA90 inducidos por las mutaciones solas o combinadas entre sí en las células endocárdicas estimuladas a 0,1-3Hz. DPA: duración del potencial de acción; WT: forma nativa del canal.")
A: potenciales de acción generados por el modelo O’Hara-Rudy en miocitos ventriculares endocárdicos a 1Hz en condiciones de control (WT) o en presencia de las mutaciones p.R1644H Nav1.5 y p.S1961N Cav1.2 por separado o combinadas. B: porcentaje de la prolongación (valores positivos) o el acortamiento (valores negativos) de la DPA90 inducidos por las mutaciones solas o combinadas entre sí en las células endocárdicas estimuladas a 0,1-3Hz. DPA: duración del potencial de acción; WT: forma nativa del canal.
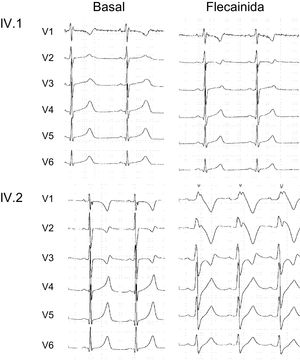
Las recomendaciones por consenso de expertos sobre las intervenciones terapéuticas en el SQTL consideran que los inhibidores de la INaL, como la flecainida, pueden ser útiles como tratamiento complementario para los pacientes con SQTL de tipo 320. En consecuencia, se comprobó la inocuidad y la eficacia de la flecainida infundiendo al paciente IV.1. el fármaco por vía intravenosa (2 mg/kg), en condiciones controladas, antes de implementar un tratamiento con este fármaco. La figura 7A muestra que, en el portador de la mutación de ganancia de función en SCN5A, la flecainida acortó el intervalo QT (el QTc se redujo de 558 a 470 ms) sin inducir respuesta arrítmica alguna. Así pues, para este chico, se añadió flecainida por vía oral al tratamiento con atenolol a la dosis máxima tolerada (50 mg/día) para la profilaxis de futuros eventos arrítmicos.
 del paciente IV.1 (arriba) y del paciente IV.2 (abajo) antes (control) y a los 10min de recibir una infusión intravenosa de flecainida 2 mg/kg.")
Por el contrario, las mutaciones de pérdida de función en el gen CACNA1C se han relacionado con el síndrome de Brugada5, cuyo diagnóstico es definitivo cuando se observa una elevación del segmento ST de tipo I en las derivaciones precordiales tras la administración de flecainida por vía intravenosa. Puesto que el análisis funcional puso de relieve que la mutación p.S1961N Cav1.2 reduce la densidad de la ICaL, la pregunta era si la presencia de la mutación en el gen CACNA1C conduciría a un patrón parecido al del síndrome de Brugada en presencia de flecainida11. En consecuencia, se realizó un test de flecainida al paciente IV.2, portador del gen SCN5A y de la mutación de pérdida de función en el gen CACNA1C. Curiosamente, la infusión intravenosa de flecainida (también a 2 mg/kg) durante 10min produjo en este paciente un ensanchamiento del QRS y un patrón parecido al del síndrome de Brugada sin modificar el QTc (figura 7).
DISCUSIÓNEn este trabajo se describe funcionalmente una nueva mutación con cambio de sentido localizada en la parte distal del dominio C-terminal de Cav1.2 (p.S1961N). Esta mutación se descubrió en un probando y en 4 miembros de una familia española con antecedentes de síncope y muerte súbita cardiaca. Los resultados demostraron que la mutación p.S1961N disminuía la densidad de la ICaL, lo que probablemente acorta la duración de la fase de meseta del PA cardiaco.
El sobrino mayor del probando sufría un SQTL de tipo 3, y se constató que era portador de la mutación p.R1644H Nav1.5. No obstante, su madre, su hermano y su tío (el probando) también eran portadores de la mutación p.R1644H Nav1.5 pero no presentaban SQTL. Esto podía explicarse por la expresividad a menudo variable del SQTL entre los portadores de una mutación, que en algunas familias puede explicarse por la presencia de múltiples mutaciones en el mismo gen u otros distintos (heterocigosis compuesta o digénica respectivamente) o por polimorfismos en un solo nucleótido (PMN)9,10,21. La compensación funcional entre mutaciones o polimorfismos en el gen SCN5A ya se ha descrito con anterioridad en algunos individuos y familias10,22,23. Es el caso del polimorfismo común p.H558R de Nav1.5 cuando aparece en el mismo alelo (cis) o en uno distinto (trans) de la mutación10,21,23. Además, se ha visto que 2 mutaciones del gen SCN5A en trans (heterocigosis compuesta) pueden interaccionar y mejorar mutuamente sus respectivos efectos deletéreos10,24. No obstante, no se hallaron otros polimorfismos o mutaciones en el gen SCN5A en ninguno de los miembros de la familia aquí estudiada.
Algunas mutaciones del gen SCN5A pueden originar un amplio espectro de fenotipos de la enfermedad entre los distintos portadores, con lo que se producen «síndromes solapados o superpuestos»11,25. De hecho, se ha demostrado que la sustitución de la Arg 1644 de los canales Nav1.5 por Cys (p.R1644C) en algunos individuos produce SQTL de tipo 326 y en otros, síndrome de Brugada27. Sin embargo, no es el caso en la familia aquí estudiada, en la que, aparte del fenotipo de SQTL de tipo 3 del paciente IV.1, no aparecieron otros rasgos electrocardiográficos que indicaran otro fenotipo arrítmico. Esto concuerda con los datos ya comentados en relación con otras familias portadoras de la mutación de ganancia de función p.R1644H, que se ha asociado exclusivamente con el SQTL de tipo 317,18.
La mutación p.S1961N de Cav1.2 reduce la densidad de la corriente de Ca de tipo LA potenciales de membrana fisiológicamente relevantes, la densidad máxima de la ICaL generada por los canales p.S1961N Cav1.2 disminuyó un ∼25% y la carga total que atraviesa la membrana, un ∼40%. Por el contrario, cuando las células se transfectaron simultáneamente con canales Cav1.2 nativos y mutantes (en proporción 0,5:0,5) para «reproducir» la condición de heterocigosis de los portadores, la disminución de la corriente se redujo. Estos resultados coinciden con el fenotipo leve-normal de los portadores de la mutación p.S1961N, cuyos ECG e intervalos QT son normales, tanto si la presentaban simultáneamente con la mutación Nav1.5 (III.1, III.2, IV.2) como si no (II.4 y III.4). Funcionalmente, el rasgo distintivo de la mutación fue la aceleración de la cinética de inactivación de los canales Cav1.2 sin que ello se acompañara de alteraciones en la dependencia del voltaje de la activación o la inactivación.
Como se ha dicho, la inactivación de los canales Cav1.2 consiste en la IDC y la IDV19,28. La IDC está controlada principalmente por la unión de Ca/calmodulina a un motivo IQ y prácticamente se anula cuando se utiliza Ba como portador de carga19. Los determinantes moleculares de la IDV, que entre otros incluyen el dominio C–terminal de los canales Cav1.2, son múltiples y complejos28. Datos previos han demostrado que la presencia de mutaciones del canal Cav1.2 en el sitio de unión a calcineurina (CaN) (residuos 1913-1941; NP_000710.5) aumenta notablemente la IDV29. Además, se ha señalado que el sitio de unión a CaN es el motivo regulador de la IDV del canal de Ca cardiaco30, que se superpone parcialmente con el sitio de unión a la proteína-fosfatasa 2A (PP2A) (residuos 1928-1970)31. Según esto, el residuo p.S1961 se ubica en la región C–terminal del sitio de unión a PP2A. Nuestros resultados han demostrado que la mutación p.S1961N aumenta principalmente la IDV de los canales Cav1.2 y, en consecuencia, podría especularse que la mutación aumenta la unión a PP2A o su función. En realidad, algunos estudios señalan que la desfosforilación mediada por la PP2A de los canales Cav1.2 inhibe su función al antagonizar el aumento de la ICaL mediado por la proteincinasa dependiente de AMPc31. Por último, la nueva mutación de pérdida de función del gen CACNA1C que se ha descrito podría eliminar, por supresión de la serina en la posición 1961, un posible sitio de fosforilación. La consideración de esta serina como un sitio posible de fosforilación para la cinasa 1 dependiente de fosfatidilinositol (PDPK1) se debe a predicciones bioinformáticas (Group-based prediction System 3.0), que no se ha demostrado experimentalmente. Por lo tanto, se requieren otros estudios para determinar el mecanismo último por medio del cual la mutación causa una aceleración de la IDV de los canales Cav1.2.
Implicaciones clínicas de los hallazgos funcionales en esta familiaLos resultados del modelo matemático confirmaron que el aumento de la INaL producido por la mutación p.R1644H de Nav1.5 podría ser el origen de la prolongación de la DPA y la duración del intervalo QT observadas en el paciente IV.1. Al contrario, la ligera disminución en la entrada de Ca durante la fase de meseta del PA cardiaco humano producido por la mutación p.S1961N del canal Cav1.2 podría compensar el aumento de la entrada de Na producido por la mutación p.R1644H del canal Nav1.5. Por consiguiente, en los miembros de la familia con ambas mutaciones, los PA ventriculares y el ECG serían «prácticamente normales». No obstante, hay que tener presente que el probando, que es portador de ambas mutaciones, sufrió un episodio sincopal cuando se hallaba en tratamiento con ciprofloxacino, un fármaco que prolonga el intervalo QT15. Además, tanto la madre como la abuela del probando, que parecerían «portadoras obligadas» de ambas mutaciones (el padre del probando no presenta ninguna de las mutaciones), sufrieron muerte súbita cardiaca. Cabe destacar que en el paciente IV.2, que también es portador de ambas mutaciones, la infusión de flecainida produjo un ensanchamiento notable del QRS e indujo un patrón electrocardiográfico parecido al del síndrome de Brugada, sin ninguna modificación en la duración del QT. En consecuencia, puede establecerse la hipótesis de que la «compensación funcional» entre mutaciones de ganancia de función y mutaciones de pérdida de función de los canales de Na y Ca se limitaría a condiciones en las que los antecedentes genéticos de los pacientes no concurren con otros factores que prolonguen la repolarización ventricular (como los fármacos, la bradicardia y las alteraciones electrolíticas) o disminuyan la excitabilidad cardiaca o acorten la duración de la fase de meseta del PA ventricular (como la flecainida). Así pues, parece que en los pacientes portadores de ambas mutaciones tanto las arritmias asociadas con taquicardia ventricular tipo torsades de pointes como las asociadas con síndrome de Brugada podrían haberse generado en función de distintos factores proarrítmicos.
CONCLUSIONESEstos resultados respaldan además la afirmación de que la penetrancia y la expresividad fenotípica variables de los síndromes arritmógenos heredados pueden atribuirse parcialmente a factores genéticos. En este contexto, el análisis funcional podría ayudar a seleccionar el tratamiento para cada miembro de la familia.
FINANCIACIÓNEste estudio fue financiado por Fondos Europeos de Desarrollo Regional, el Ministerio de Economía y Competitividad [SAF2014-58769-P; SAF2017-88116-P], la Comunidad Autónoma de Madrid [B2017/BMD-3738], el Instituto de Salud Carlos III [PI16/00398], la Red ERA para Programas de Investigación en Enfermedades Raras (AC14/00029), las fundaciones de la Mutua Madrileña y del Banco Bilbao Vizcaya Argentaria y la Sociedad Española de Cardiología.
CONFLICTO DE INTERESESNo se declara ninguno.
- –
Por lo menos son 15 los genes vinculados hasta ahora con el SQTL, un síndrome arritmogénico primario que se caracteriza por un intervalo QT largo que aumenta el riesgo de muerte súbita cardiaca debida a fibrilación ventricular. El SQTL muestra una notable expresividad fenotípica, que se ha atribuido principalmente a factores demográficos y dificulta enormemente la estratificación del riesgo en los pacientes y las decisiones terapéuticas. No obstante, en algunas familias, la expresividad variable se debe a factores genéticos y puede explicarse por la presencia de múltiples mutaciones en el mismo gen o distintos genes (heterocigosis compuesta o digénica, respectivamente) o por la presencia de polimorfismos.
- –
Se analizó funcionalmente una mutación del gen CACNA1C, identificada en heterocigosis digénica en una familia española, que codifica los canales p.S1691N Cav1.2. Los resultados pusieron de relieve que esta mutación disminuía la entrada de Ca durante la fase de meseta del PA, efecto que contrarresta funcionalmente el aumento de entrada de Na producido por la mutación del gen SCN5A asociada con QT largo que codifica los canales p.R1644H Nav1.5. Como resultado, solo el miembro de la familia que portaba exclusivamente la mutación en el gen SCN5A tiene SQTL de tipo 3. Así pues, la expresión fenotípica del SQTL podría modularse por medio de factores genéticos, y el análisis funcional de mutaciones concurrentes podría servir para orientar un enfoque terapéutico personalizado.
A Sandra Sacristán, Lorena Ondo y Paloma Vaquero por su inestimable ayuda técnica.