


La hipertensión pulmonar (HP) se define como la elevación de la presión pulmonar media ≥ 25 mmHg en reposo. En los pacientes adultos con cardiopatías congénitas puede producirse tanto por afección del corazón izquierdo con elevación de la presión poscapilar como por un shunt sistémico-pulmonar que induce la elevación de la presión precapilar. En este artículo se revisará el segundo grupo, desarrollando las causas, los mecanismos implicados y los tratamientos actuales. Se prestará especial atención al manejo postoperatorio de la HP en los pacientes con shunt sistemicopulmonares y los métodos utilizados para discriminar entre la HP severa fija e irreversible y la HP con distintos grados de severidad pero potencialmente reversible, que permite la corrección del cortocircuito. Asimismo, se expondrán los recientes avances en la clasificación de la HP en relación con cardiopatías congénitas y el algoritmo terapéutico recomendado.
Palabras clave
La hipertensión pulmonar (HP) es un factor implicado en el pronóstico y la supervivencia de los adultos con cardiopatía congénita. El incremento de las presiones pulmonares se desarrolla en dos contextos diferentes: a) las cardiopatías congénitas (CC) que afectan al corazón izquierdo y producen una elevación de la presión poscapilar, y b) las CC que producen un cortocircuito izquierda-derecha intracardiaco o extracardiaco e inducen una elevación de la presión precapilar. En esta revisión nos centraremos en el segundo grupo, ya que el manejo de la HP poscapilar en las CC no difiere sustancialmente del de la HP poscapilar en las cardiopatías adquiridas.
En los pacientes con CC, el incremento del flujo y la presión intravascular producen un exceso del estrés tangencial o de cizallamiento (shear stress) en la pared vascular del lecho pulmonar que desencadenan disfunción del endotelio vascular y liberación de sustancias vasoactivas. Se produce un déficit de óxido nítrico y prostacilina, junto con un incremento de endotelina 1 y tromboxano A2, que determina un predominio del tono vasoconstrictor y la hipercoagulabilidad y, fundamentalmente, incrementa la proliferación celular. El resultado es un remodelado vascular que afecta a todas la capas de la pared del vaso, incrementa su grosor, disminuye su distensibilidad y oblitera la luz. Los cambios histopatológicos que se producen son progresivos, de forma que en las fases iniciales el remodelado vascular es reversible (hipertrofia de la media e hiperplasia de la íntima) si se solventa el hiperaflujo pulmonar. Sin embargo, si la situación persiste, se produce una obliteración del lecho vascular pulmonar con la aparición de lesiones plexiformes y arteritis consideradas irreversibles.
CLASIFICACIÓN DE LAS CARDIOPATÍAS CONGÉNITAS CON SHUNT SISTÉMICO PULMONARLa clasificación propuesta (tabla 1) es una modificación1 de la realizada en el Tercer Congreso Mundial de HP, celebrado en Venecia en 20032. Se consideran cuatro factores que condicionan el diagnóstico, el pronóstico y el tratamiento de los pacientes con HP y CC:
- –
Tipo: las CC que producen HP pueden ser simples o complejas. El establecimiento de la enfermedad obstructiva del vaso pulmonar depende del tamaño y la localización del cortocircuito, así como del grado de sobrecarga de volumen y presión que produce. En los defectos pretricuspídeos, la incidencia de HP es menor que en los postricuspídeos, ya que sólo se produce sobrecarga de volumen frente a la sobrecarga de presión y volumen de los últimos.
- –
Tamaño: la importancia del tamaño en el shunt tiene dos componentes: la dimensión del defecto y el comportamiento hemodinámico (gradiente de presión a través del shunt). Así un shunt se considera pequeño si tiene dimensiones reducidas y es restrictivo.
- –
Dirección del shunt: nos da una información fundamental. En el síndrome de Eisenmenger (SE) la dirección es derecha a izquierda o bidireccional, en las otras formas de HP asociada a CC el shunt es izquierda-derecha.
- –
Lesiones asociadas: hay alteraciones extracardiacas que influyen en la aparición, la precocidad y la severidad de la HP; una de las más relevantes es el síndrome de Down.
- –
Estado de la reparación: el comportamiento de la HP es diferente en los pacientes con SE que en los pacientes con HP severa tras la cirugía.
Clasificación anatomopatológica de los shunts sistemicopulmonares congénitos asociados a hipertensión pulmonar
|
CIA: comunicación interauricular; CIV: comunicación interventricular.
Esta clasificación intenta describir con detalle las variables anatómicas, fisiológicas y hemodinámicas que condicionan la aparición de HP en las CC, pero puede resultar muy complicada en la práctica diaria. Desde un punto de vista clínico, se pueden definir cuatro grupos bien diferenciados (tabla 2):
Clasificación clínica de los shunts sistemicopulmonares congénitos asociados a hipertensión pulmonar
|
RVP: resistencias vasculares pulmonares.
Incluye la mayor parte de los defectos grandes distales a la válvula tricúspide. Se establece en los primeros años de vida y no es subsidiario de ningún procedimiento de reparación.
En la mayoría de los pacientes con amplia comunicación sistemicopulmonar, las resistencias vasculares pulmonares disminuyen tras el nacimiento, produciendo un incremento del shunt izquierda-derecha. Los síntomas de insuficiencia cardiaca secundarios al hiperaflujo pulmonar y la cianosis en el ejercicio son propios de esta fase. El incremento de flujo y la presión intravascular producen un exceso del estrés tangencial que desencadena disfunción del endotelio vascular y liberación de sustancias vasoactivas que dan lugar a vasoconstricción y remodelado vascular. Estos cambios contribuirán a un incremento progresivo de las resistencias vasculares pulmonares (RVP). Los síntomas de insuficiencia cardiaca secundarios a un estado hiperdinámico desaparecen y la cianosis está presente en reposo, instaurándose el SE.
El SE afecta a múltiples órganos cuya función se deteriora con el tiempo (tabla 3). Se consideran marcadores de mal pronóstico la aparición de arritmias, la desaturación sistémica progresiva y la insuficiencia cardiaca. La supervivencia es claramente menor que la de la población general (sólo el 50% alcanza la edad de 60 años, y la supervivencia es menor en los pacientes con CC complejas), pero mejor que la de los pacientes con HP idiopática en la misma clase funcional3. En una serie de 100 pacientes listados para trasplante, la supervivencia de los no trasplantados fue del 97% al año, el 89% a los 2 años y el 77% a los 3 años en el SE y del 77, el 69 y el 35%, respectivamente, en la HP idiopática4.
Características clínicas del síndrome de Eisenmenger
| Anomalías | Manifestaciones clínicas |
| Elevación severa de la resistencia vascular pulmonar | Intolerancia al ejercicio, disnea, síncope, muerte súbita |
| Eritrocitosis secundaria | Hiperviscosidad, deficiencia de vitamina B12, ácido fólico y hierro |
| Diátesis hemorrágica | Hemoptisis, hemorragia cerebral, metrorragia, epistaxis |
| Fallo del ventrículo derecho | Hepatomegalia, edema |
| Arritmias | Síncope, muerte súbita |
| Alteraciones hemáticas y diátesis trombótica | Accidentes cerebrovasculares, trombosis en las arterias pulmonares |
| Disfunción renal | Aumento del nitrógeno de la urea en sangre, hiperuricemia y gota |
| Disfunción hepaticobiliar | Litiasis y colecistitis |
| Infecciones | Endocarditis, absceso cerebral |
| Alteraciones óseas | Escoliosis y osteoartropatía hipertrófica |
Habitualmente de localización pretricuspídea, presenta HP severa pero mantiene cortocircuito izquierda-derecha (saturación periférica de O2 > 90%) y podría considerarse una situación transitoria que conduce al SE. Sin embargo, en la edad adulta, al contrario que en la infancia, es frecuente que se mantenga esta situación de HP hipercinética sin que se llegue a completar la evolución hacia una fisiología de SE. La evolución natural de este grupo de pacientes es peor que la del SE. Así, en un estudio realizado en 60 pacientes, 41 con SE y 19 con HP hipercinética, el 80% de los pacientes con SE estaban vivos a los 20 años del diagnóstico frente a un 20% en el grupo de HP hipercinética5. En estos pacientes la reparación de la cardiopatía es eficaz para suprimir el cortocircuito, y si las presiones pulmonares se normalizan tras la intervención, mejora el pronóstico y la calidad de vida. Sin embargo, si la HP persiste tras la reparación, la supervivencia disminuye drásticamente, siendo claramente menor que la HP hipercinética no reparada. En este grupo de pacientes, es fundamental el estudio de la reversibilidad de la HP antes de la actuación sobre el shunt sistemicopulmonar.
En general, se considera que un índice de resistencias vasculares pulmonares (IRVP) < 6 UW/m2 indica HP reversible subsidiaria de corrección si el Qp/Qs es > 1,5. Si las RVP son más elevadas requiere un estudio más minucioso de la reversibilidad de la HP. Para valorar la reversibilidad de la HP en los pacientes con CC se utilizan fundamentalmente dos procedimientos:
- 1.
El test agudo vasodilatador. Según el consenso realizado por el instituto de investigación vascular pulmonar para el manejo de HP en CC6, se considera que los pacientes pueden ser candidatos a la reparación si se produce una reducción del 20% del IRVP y del cociente de resistencias pulmonares/sistémicas, con unos valores finales de IRVP < 6 UW/ m2 y un cociente de resistencias < 0,3. Estos límites no están claramente establecidos y son modificables según la edad del paciente y el tipo de CC. En general, se es más permisivo con los defectos pretricuspídeos y más restrictivo a mayor edad del paciente. Los fármacos más utilizados son el oxígeno y el óxido nítrico (una mezcla compuesta por 80–20ppm de óxido nítrico más el 100% de oxígeno durante 10min).
- 2.
Oclusión temporal del defecto. Se fundamenta en analizar la respuesta de la circulación pulmonar a la oclusión temporal en la sala de hemodinámica del shunt izquierda-derecha (comunicación interauricular [CIA] tipo ostium secundum o ductus arterioso persistente). Tras la oclusión del defecto, se considera respuesta favorable el descenso > 25% de las presiones pulmonares o la reducción > 50% del cociente entre la presión diastólica pulmonar y la presión diastólica aórtica, sin datos de claudicación del ventrículo derecho (VD) (incrementos de la presión en aurícula derecha [AD] y telediastólica de VD) ni disminución de la presión arterial sistémica o elevación de la presión capilar pulmonar7. Estudios preliminares consideran este «ensayo terapéutico» seguro y buen predictor del comportamiento de la HP tras la reparación del cortocircuito, si bien en el momento actual la experiencia es escasa.
El comportamiento clínico es muy similar al descrito en la HP idiopática; si hay cianosis, ésta es leve y sin afección secundaria de otros órganos. Es una situación de solapamiento entre la HP idiopática y la HP asociada a shunt sistemicopulmonar y el papel del shunt en el desarrollo de la HP no está bien establecido.
La HP tras una cirugía correctivaSe distinguen dos estadios:
- –
La HP en el postoperatorio de la cirugía cardiaca. Es una de las complicaciones que producen mayor morbimortalidad. En una revisión de las series recientes8, la HP complica el postoperatorio del 2% de los pacientes con CC y las crisis de HP se producen en el 0,75%. Los pacientes con mayor riesgo de presentar crisis de HP son los que tienen un lecho pulmonar muy reactivo, síndrome de Down o elevación de la presiones en la aurícula izquierda. Además, los pacientes con disfunción ventricular derecha y severa regurgitación tricuspídea toleran mal incluso grados moderados de HP. Ambos grupos se benefician del tratamiento precoz con vasodilatadores pulmonares. El manejo postoperatorio de estos pacientes va dirigido a disminuir las RVP y preservar la función del VD. Para ello, se utilizan distintas estrategias: a) optimizar la ventilación, manteniendo una baja presión en la vía aérea e hiperventilación con el objetivo de incrementar el pH hasta el límite alto de la normalidad, manteniendo el CO2 en sus límites inferiores; b) soporte inotrópico, evitando en lo posible los fármacos que producen estímulo alfa y vasoconstricción, y c) si se producen crisis hipertensivas o las RVP permanecen elevadas y comprometen la función del VD, el siguiente escalón terapéutico es el óxido nítrico (NO); en general, se necesitan bajas concentraciones (20ppm). En algunos pacientes con RVP severamente elevadas y que han requerido dosis altas de NO, no es posible suspender su administración por la aparición de HP de rebote y se ha utilizado con éxito la administración de sildenafilo vía oral, lo que permite suspender el NO con control adecuado de la HP9. La utilización de iloprost inhalado (análogo estable de la prostaciclina) puede ser también una alternativa eficaz en el manejo de estos pacientes con RVP altas e insuficiencia cardiaca10.
- –
La HP crónica tras la cirugía cardiaca. En ésta podemos encontrar dos grupos de pacientes, los que tienen HP persistente tras la cirugía o los que la desarrollan tras una cirugía con éxito sin HP significativa previa. El tiempo de aparición de HP severa tras la cirugía es muy variable.
En el registro español de HP (REHAP)11, la prevalencia estimada de HP severa asociada a CC es de 3,1 casos por millón de habitantes mayores de 14 años (el 16% de los pacientes del registro tienen HP asociada a CC). El 32% de los pacientes tienen HP tras cirugía; el 35%, HP asociada a shunt pretricuspídeo y el 78%, asociada a CC simple. El desarrollo de HP en 1.897 pacientes adultos con shunt sistemicopulmonares se objetivó en un 28%, según los datos del Euro Heart Survey Registry12; sin embargo, en el registro holandés, con 1.824 pacientes, sólo el 6% desarrollaba HP13. Si consideramos todos estos datos de forma conjunta, podríamos estimar una prevalencia de HP asociada a CC en Europa de 1,6 a 12,5 casos por millón de habitantes en edad adulta2.
TRATAMIENTO MÉDICOLa HP asociada a CC pertenece al grupo 1 de la clasificación del Tercer Congreso Mundial de HP, celebrado en Venecia en 2003, y estos pacientes comparten similitudes biopatológicas y estructurales con los pacientes con HP idiopática o asociada a otras entidades. Por ello, los objetivos del tratamiento se dirigen a las vías moleculares alteradas en esta enfermedad, como la del óxido nítrico (inhibidores de la fosfodiesterasa 5, NO), la de la endotelina (antagonistas de los receptores de la endotelina) y la de la prostaciclina (epoprostenol y análogos), esperando obtener un efecto beneficioso vasodilatador y antiproliferativo. Algunos de los grandes ensayos clínicos en HP, llevados a cabo con los fármacos descritos previamente, incluyeron a pacientes con HP asociada a CC, aunque no hay datos publicados de este subgrupo específico y se extrapolan de los resultados generales. En la actualidad hay un único estudio aleatorizado controlado con placebo realizado con bosentán14, y los niveles de evidencia para el uso del resto de los fármacos se basan en la experiencia clínica de centros expertos.
Los pacientes con HP asociada a CC con defectos de pequeño tamaño y con HP persistente tras corrección quirúrgica sin shunt residual se deben tratar de forma similar a aquellos con HP idiopática15. En este apartado se hablará específicamente de la HP asociada a shunt sistemicopulmonar no restrictivo y del SE.
Antagonistas de los receptores de la endotelinaLa endotelina 1 tiene un papel importante en las alteraciones funcionales y estructurales y en el desarrollo de la HP asociada a CC. Estudios abiertos no controlados y series de casos con bosentán16, antagonista dual de los receptores de endotelina, apuntaron a un perfil de seguridad en cuanto a la desaturación sistémica y una mejora funcional y hemodinámica en pacientes con SE. El BREATHE-5 (Bosentan Randomised trial of Endothelin Antagonist THErapy)14 es el único estudio doble ciego, aleatorizado, controlado con placebo, llevado a cabo hasta el momento en pacientes con SE en clase funcional III de la NYHA. En este estudio, se incluyó a 54 pacientes y el tratamiento con bosentán demostró una mejora significativa de la capacidad de ejercicio (+34m en el test de la marcha de 6min) y una reducción del índice de RVP (−472dyn/s/cm5), sin desaturación sistémica, a las 16 semanas de seguimiento. En el estudio de extensión, a las 40 semanas se mantuvo el efecto beneficioso del fármaco17. En un reciente subanálisis, en el que se diferencian los subgrupos de pacientes con comunicación interventricular o con CIA, no se observaron diferencias en la eficacia del tratamiento ni en el grado de desaturación sistémica18. Los resultados de estudios observacionales a más largo plazo (al menos 2 años) indican el efecto beneficioso sostenido en la mejora de la clase funcional y el perfil de seguridad del fármaco19.
El sitaxentán es un antagonista selectivo de los receptores tipo A de la endotelina. En el estudio STRIDE-2 se incluyó a un 15% de los pacientes con HP asociada a CC. No se realizó un subanálisis específico de este grupo de pacientes, aunque globalmente mejoraron la capacidad de ejercicio y la clase funcional20. En un reciente análisis retrospectivo de un grupo de 14 pacientes tratados con sitaxentán, se observó mejora de las RVP sin empeoramiento del shunt D-I21.
Inhibidores de la fosfodiesterasa tipo 5El sildenafilo y el tadalafilo son potentes inhibidores de la fosfodiesterasa tipo 5, que incrementan los valores de GMPc y ejercen un efecto beneficioso produciendo vasodilatación selectiva en la circulación pulmonar. Hasta la fecha, se han realizado pocos estudios, con escaso número de pacientes. En un estudio abierto prospectivo se observó la evolución favorable de 7 pacientes con SE tratados con sildenafilo durante 6 meses con mejora de la clase funcional, la saturación sistémica de O2 (reducción de la cianosis) y los parámetros hemodinámicos, sin efectos adversos22. En otro estudio observacional, se administró tadalafilo durante 3 meses a 16 pacientes con SE, y se observó un efecto beneficioso en la capacidad funcional y las RVP23.
Prostaciclina y análogosEl epoprostenol intravenoso se ha utilizado en diversos estudios no controlados en pacientes con HP asociada a CC, y ha mostrado un efecto beneficioso a largo plazo (1 año de seguimiento) en términos de clase funcional y severidad de los parámetros hemodinámicos24. Sin embargo, la administración de epoprostenol está limitada por las serias complicaciones que produce, como la infección, la trombosis, la embolia paradójica en pacientes con shunt sistemicopulmonar o el desplazamiento del catéter, todas ellas potencialmente fatales.
La eficacia del treprostinil administrado por vía subcutánea fue evaluada en el estudio llevado a cabo por Simonneau et al25. En ese estudio, 109 pacientes presentaban HP asociada a shunt sistemicopulmonar, SE y HP persistente tras corrección quirúrgica. Aunque no se han publicado los datos del subgrupo de los pacientes con CC, se describió un efecto beneficioso en la población total del estudio.
Hay una experiencia muy limitada con iloprost inhalado a largo plazo en los pacientes con CC, y su uso principal se ha dirigido al manejo postoperatorio de la HP en la cirugía de las CC.
Tratamiento combinadoEn la actualidad, el tratamiento combinado se utiliza en la mayoría de los centros de referencia cuando en monoterapia no se alcanzan los objetivos terapéuticos. La base racional para su uso es buscar los efectos sinérgicos y aditivos actuando sobre diferentes mecanismos patogénicos y reduciendo al mínimo la toxicidad. Existen múltiples estudios observacionales y controlados que demuestran el beneficio de la terapia combinada en los pacientes con HP15. Sin embargo, no hay hasta la fecha ningún estudio que incluya a pacientes con HP asociada a CC y su uso se sustenta en prácticas clínicas favorables en centros con experiencia.
Tratamiento médico específico como puente a una posible reparación del defectoEl tratamiento médico específico consigue, en algunos casos, una reducción significativa de las RVP y plantea la posibilidad de la reparación del shunt en pacientes en quienes inicialmente se ha desestimado por la severidad de la HP. Se han descrito varias series de casos clínicos26 en los que se ha conseguido reducir las RVP, la mayor parte en pacientes con CIA, lo que hace posible la reparación del defecto anatómico con buena evolución a corto plazo. Hoy en día, aunque estas publicaciones anecdóticas son prometedoras, se desconoce qué pacientes podrían beneficiarse, y son necesarios estudios multicéntricos, con largo seguimiento, que permitan establecer la seguridad y la eficacia de este procedimiento.
Trasplante cardiopulmonarEl trasplante cardiopulmonar o el pulmonar con corrección anatómica del defecto cardiaco son opciones terapéuticas en pacientes seleccionados con síndrome de Eisenmenger, y han demostrado mejorar la calidad de vida27. En la práctica, se restringe a pacientes muy sintomáticos, con resistencia a otro tipo de terapias farmacológicas y con criterios de mal pronóstico a corto plazo (síncope, insuficiencia cardiaca resistente, clase funcional avanzada e hipoxemia severa).
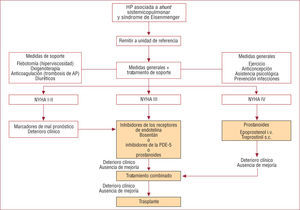
ALGORITMO TERAPÉUTICOEl algoritmo terapéutico (fig. 1) está basado en la opinión de expertos1,28, no está sustentado en la evidencia científica y, por ello, no se establece graduación en el nivel de recomendación.

Algoritmo terapéutico. Modificada de Galie et al1. AP: arteria pulmonar; HP: hipertensión pulmonar; i.v.: intravenoso; PDE: fosfodiesterasa; s.c.: subcutáneo.
Las medidas generales deben aplicarse a todos los pacientes e incluyen recomendaciones sobre la actividad física (evitar temperaturas extremas y deshidratación), la profilaxis de la endocarditis bacteriana, el tratamiento precoz de las infecciones y la vacunación contra la gripe y el neumococo. El uso de oxígeno suplementario es controvertido y se recomienda en los pacientes en que se objetiva un incremento de la saturación periférica de O2 al administrarlo. La indicación de anticoagulación no está bien establecida; en general, se recomienda en presencia de arritmias auriculares, insuficiencia cardiaca y trombosis de las arterias pulmonares en ausencia de hemoptisis grave. Asimismo, se debe intentar preservar el ritmo sinusal siempre que sea posible (fármacos antiarrítmicos y/o cardioversión eléctrica); la cirugía no cardiaca debe evitarse, si es posible, y siempre requiere una planificación cuidadosa. La anestesia epidural es de elección y se debe sustituir los fármacos orales, si se contempla un intervalo de ayuno superior a las 24h, por epoprostenol intravenoso. El embarazo, la anticoncepción y la flebotomía son tratados en otros artículos de esta monografía.
El tratamiento con fármacos específicos de la HP se considera en los pacientes en clase funcional III e incluye antagonistas de los receptores de la endotelina, inhibidores de la fosfodiesterasa 5 y prostaciclina. El tratamiento combinado y el trasplante están indicados en los pacientes gravemente enfermos y con fracaso de la monoterapia.
CONCLUSIONESLos pacientes con HP asociada a CC tienen características diferenciales: la presencia del shunt, la complejidad de la cardiopatía subyacente, la excelente adaptación del ventrículo derecho, la cianosis y el deterioro multiorgánico. Además, presentan mejor pronóstico y mayor supervivencia que aquellos con otras formas de HP. Los fármacos utilizados en el tratamiento de otras formas de HP también parecen ser eficaces en estos pacientes; sin embargo, no se conoce bien si el efecto beneficioso se mantiene a largo plazo. Actualmente, únicamente el bosentán (antagonista dual de los receptores de la endotelina) ha demostrado su eficacia y seguridad en el tratamiento de los pacientes con SE en un ensayo clínico. Se ha propuesto un algoritmo terapéutico para los pacientes con shunt sistemicopulmonar no restrictivo y para el SE adaptado del algoritmo terapéutico de la HP idiopática.