La miocardiopatía no compactada se caracteriza por la presencia de un ventrículo con marcadas trabeculaciones. Aunque su origen se ha asociado con la detención de la compactación normal del miocardio durante el primer trimestre de gestación1, también puede tratarse de un rasgo adquirido en determinadas situaciones fisiológicas (deporte, embarazo) o patológicas (hipertensión, anemia) o en relación con otras miocardiopatías y cardiopatías congénitas. El diagnóstico se basa en las pruebas de imagen2 y el paciente puede estar asintomático o presentar clínica de insuficiencia cardiaca, arritmias o secundaria a fenómenos tromboembólicos. Factores como el grado de disfunción y dilatación ventricular y la presencia de captación tardía en la cardiorresonacia se han relacionado con peor evolución3. Se ha descrito disfunción ventricular grave al diagnóstico, con recuperación parcial o completa de la función y empeoramiento posterior4.
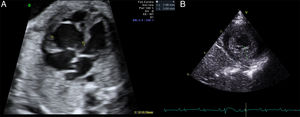
Se presenta el caso de una neonata con diagnóstico prenatal de miocardiopatía no compactada. La madre tenía el antecedente de óbito fetal por hidropersía de causa no filiada en una gestación previa. A las 28 semanas de gestación, se observó un ventrículo derecho hipertrabeculado con disfunción grave (figura A), ausencia de flujo anterógrado pulmonar, insuficiencia tricuspídea grave y un ventrículo izquierdo no compactado con función conservada. Se inició tratamiento materno con digoxina hasta la semana 36 de gestación, y en el ecocardiograma se observó mejoría de la función ventricular derecha.

Nació a las 39 semanas de gestación, con buena situación clínica, y el ecocardiograma al ingreso mostraba trabeculaciones en el ventrículo izquierdo (figura B) con disfunción ventricular izquierda grave y derecha moderada (tabla). El electrocardiograma mostraba una alteración de la repolarización con ondas T negativas de V3 a V6. Se inició tratamiento con milrinona y, a los 8 días de vida, presentó discreta mejoría de la función ventricular. Se le dio el alta en tratamiento con inhibidores de la enzima de conversión de la angiotensiona y bloqueadores beta.
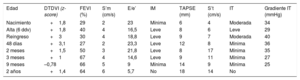
Evolución ecocardiográfica
| Edad | DTDVI (z-score) | FEVI (%) | S’m (cm/s) | E/e’ | IM | TAPSE (mm) | S’t (cm/s) | IT | Gradiente IT (mmHg) |
|---|---|---|---|---|---|---|---|---|---|
| Nacimiento | +1,8 | 29 | 2 | 23 | Mínima | 6 | 4 | Moderada | 34 |
| Alta (6 ddv) | +1,8 | 40 | 4 | 16,5 | Leve | 8 | 6 | Leve | 29 |
| Reingreso | +3 | 30 | 4 | 18,8 | Leve | 9 | 7 | Moderada | 40 |
| 48 días | +3,1 | 27 | 2 | 23,3 | Leve | 12 | 8 | Mínima | 36 |
| 2 meses | +1,5 | 50 | 3 | 21,8 | Leve | 8 | 17 | Mínima | 35 |
| 3 meses | +1 | 67 | 4 | 14,6 | Leve | 9 | 11 | Mínima | 27 |
| 9 meses | –0,78 | 66 | 5 | 9 | Mínima | 14 | 9 | Mínima | 25 |
| 2 años | +1,4 | 64 | 6 | 5,7 | No | 18 | 14 | No |
ddv: días de vida; DTDVI: diámetro telediastólico del ventrículo izquierdo; FEVI: fracción de eyección del ventrículo izquierdo; IM: insuficiencia mitral; IT: insuficiencia tricuspídea; S’m: S tisular mitral; S’t: S tisular tricuspídea; TAPSE: desplazamiento sistólico del plano del anillo tricuspídeo.
A los 34 días de vida, sufrió un shock cardiogénico y requirió asistencia respiratoria y hemodinámica. La ecocardiografía mostraba una disfunción ventricular izquierda grave con hipertensión pulmonar de causa retrógrada y función ventricular derecha conservada (tabla). Se mantuvo la asistencia hemodinámica y respiratoria hasta el décimo día de ingreso. Se reinició su tratamiento de base y se añadió digoxina. Se valoró la posibilidad de inclusión en lista de trasplante cardiaco, que se desestimó ante la mejoría clínica y ecocardiográfica de la paciente a partir de los 2 meses de vida, con posterior normalización de la función ventricular (tabla). Tras un seguimiento de 2 años, la paciente estaba asintomática y la ecocardiografía mostraba una función sistólica y diastólica biventricular normalizada. Durante la evolución, no se han presentado fenómenos arrítmicos ni embólicos. Ante los casos descritos con mejoría de la función y posterior empeoramiento, se decidió mantener el tratamiento con inhibidores de la enzima de conversión de la angiotensina y bloqueadores beta.
En cuanto a la etiología, se realizó estudio cardiológico de los padres y la hermana, que fue normal, y estudio genético de todos ellos. En la paciente se identificaron 2 mutaciones en el gen LDB35, ambas descritas previamente como patogénicas en casos de miocardiopatía no compactada. El estudio genético de la hermana fue normal y el de los padres mostró una mutación en cada uno de ellos.
Como conclusiones, el diagnóstico de la miocardiopatía no compactada se puede realizar en periodo intrauterino. Durante la vida fetal es frecuente que se presente con disfunción ventricular derecha, ya que es este el ventrículo dominante. Tras el nacimiento, con la caída de las resistencias vasculares pulmonares y el incremento de las sistémicas, es frecuente la mejoría de la función derecha y la aparición de disfunción ventricular izquierda. En cuanto al tratamiento, algunos autores apuntan que la introducción de un tratamiento médico precoz puede mejorar la evolución y que la respuesta a este podría ser mayor que la observada en otras miocardiopatías6. Por todo ello y ante la posibilidad ya descrita de un empeoramiento durante la evolución4, se ha mantenido el tratamiento médico a pesar de la recuperación de la función. En cuanto al estudio familiar, es recomendable realizar una valoración cardiológica de todos los familiares de primer grado. Las recomendaciones sobre cuándo realizar un estudio genético no son claras. Ante un caso aislado de miocardiopatía no compactada, este es positivo solo en un 15-40% de los casos, según las distintas publicaciones. Creemos que el estudio genético es una parte importante en la valoración de estos pacientes y que se trata de una herramienta fundamental para obtener un mayor grado de conocimiento sobre esta enfermedad. El descubrimiento de nuevos genes implicados y la progresiva disminución de los costes harán que este acabe formando parte de la práctica clínica habitual.