Palabras clave
INTRODUCCIÓN
La insuficiencia cardiaca es la enfermedad cardiovascular más costosa en países industrializados y la primera causa de hospitalización en adultos mayores de 60 años. A pesar de los avances terapéuticos, tras su diagnóstico el riesgo de muerte al año es del 20-50% según la población. Por todo esto, hay una necesidad evidente de nuevas estrategias que puedan alterar la evolución de la enfermedad, aliviar los síntomas y prolongar la vida del paciente. La apoptosis es una de las causas principales de la muerte de cardiocitos y de la progresión de la insuficiencia cardiaca1. La posibilidad de disminuir la pérdida de cardiocitos al inhibir la apoptosis supone una importante vía de investigación para mejorar los tratamientos actuales de la insuficiencia cardiaca.
Varios trabajos han demostrado que en varones con insuficiencia cardiaca es frecuente la insuficiencia de hormonas anabólicas, y que las deficiencias de factor de crecimiento insulínico tipo 1 (IGF-1) y testosterona se asocian a un peor pronóstico y mayor mortalidad2-4. Estudios con cultivos de cardiocitos H9c2 han demostrado que la IGF-1, mediante un mecanismo rápido y no genómico, es capaz de proteger a los cardiocitos de la apoptosis inducida por estrés hiperosmótico5. En células del músculo esquelético y neuronas, la testosterona también protege de la muerte celular por un mecanismo independiente del receptor de andrógenos6,7, es decir, independiente de la ruta genómica clásica establecida para la testosterona. Actualmente se sabe que la testosterona estimula la contractilidad cardiaca, induce vasodilatación directa y mejora la función endotelial periférica8, pero no se han publicado estudios sobre el posible papel de la testosterona como modulador de la apoptosis en los cardiomiocitos.
Por otro lado, espironolactona y eplerenona son dos fármacos que han demostrado mejorar el pronóstico en pacientes con insuficiencia cardiaca severa9,10. Ambos fármacos son antagonistas de la aldosterona, pero presentan notables diferencias estructurales: la espironolactona es un antimineralocorticoide al que se atribuyen efectos progestágenos y antiandrogénicos; la eplerenona es un derivado de la espironolactona al que se han minimizado dichos efectos progestágenos y antiandrogénicos para potenciar su unión selectiva al receptor de aldosterona11,12. Estas diferencias de selectividad entre ambos fármacos podrían derivar en un comportamiento diferente en el posible papel protector de la testosterona descrito anteriormente.
El objetivo de este trabajo es estudiar si la testosterona protege de la apoptosis a los cardiocitos, sus posibles mecanismos y si la espironolactona y la eplerenona pueden tener un comportamiento diferente en dicho efecto beneficioso de la testosterona.
MÉTODOS
Diseño
Se estudiaron los efectos de la testosterona y el tratamiento combinado con espironolactona o eplerenona en la apoptosis inducida por la adición de sorbitol en cultivos celulares de cardiocitos embrionarios de rata de la línea H9c2. El sorbitol induce estrés hiperosmótico, y éste es uno de los principales mecanismos de daño tisular en estados patológicos como la isquemia, el choque séptico y la acidosis. Se valoró el posible efecto protector de la testosterona contra la apoptosis inducida y luego el efecto de la adición de bloqueadores de los receptores de aldosterona, eplerenona y espironolactona. La apoptosis se determinó mediante análisis de viabilidad celular, fragmentación del ADN y activación de las caspasas 3, 8 y 9. Además, se administró flutamida, un antagonista del receptor de andrógenos, con el fin de estudiar la implicación del receptor de andrógenos, y se analizó la participación de las rutas de señalización SAPK/JNK, ERK1/2 y p-38 proteincinasa activada por mitógenos (MAPK) para estudiar la posible implicación de mecanismos no genómicos.
Reactivos
El sorbitol, la testosterona, la flutamida y los demás reactivos, a menos que se especifique otra cosa, se obtuvieron de Sigma-Aldrich Corp. (St. Louis, Missouri, Estados Unidos). Pfizer Inc. (New York, New York, Estados Unidos) suministró la eplerenona y la espironolactona. Los reactivos necesarios para el cultivo celular como medio de cultivo (DMEM), L-glutamina, suero fetal bovino, penicilina, estreptomicina y tripsina/EDTA, se obtuvieron de Gibco (Invitrogen, Carlsbad, California, Estados Unidos). Los anticuerpos contra alfaactina y anti-IgG de conejo o ratón conjugados con peroxidasa de rábano los suministró Santa Cruz Biotechnology (Santa Cruz, California, Estados Unidos). Cell Signaling Technology, Inc. (Danvers, Massachusetts, Estados Unidos) suministró los anticuerpos contra las formas fosforiladas y total de SAPK/JNK, p38 y ERK1/2, así como contra las caspasas 3, 8 y 9. Los reactivos de detección ECL, películas autorradiográficas y los marcadores de peso molecular se obtuvieron de Amersham Pharmacia Biotech (Piscataway, New Jersey, Estados Unidos).
Cultivo celular y tratamientos
La línea celular H9c2, obtenida a partir de corazones de ratas embrionarias, fue cedida por el Dr. F. Fernández Belda (Departamento de Bioquímica, Universidad de Murcia). Las células H9c2 se mantuvieron en cultivo siguiendo protocolos estándar, a 37 °C en atmósfera húmeda con el 95% de aire y el 5% de CO2 y en medio de cultivo DMEM suplementado con 10% de suero fetal bovino, L-glutamina 2 mM, penicilina 100 U/ml y estreptomicina 100 μg/ml. Todos los tratamientos se realizaron en ausencia de suero fetal bovino y antibióticos. La apoptosis se indujo mediante estrés hiperosmótico con sorbitol (3 h, 0,6 M). La testosterona (100 nM) se administró simultáneamente con el sorbitol, a menos que se indique otra cosa. La flutamida (10 μM), la espironolactona (10 μM) y la eplerenona (10 μM) se administraron 30 min antes del tratamiento con sorbitol y testosterona. La dosis de flutamida elegida ha sido previamente validada por otros investigadores13, mientras que las dosis de espironolactona y eplerenona fueron validadas mediante ensayos con concentraciones entre 100 nM y 100 μM, escogiendo la concentración mayor con que el fármaco no tuvo efecto propio en la viabilidad celular o la activación de las caspasas (datos no mostrados).
Determinación de viabilidad celular
El porcentaje de células vivas se determinó mediante la técnica de exclusión de azul tripán. De forma resumida, una vez realizados los tratamientos, la células se lavaron en tampón fosfato (PBS) y se resuspendieron con tripsina/EDTA; una vez detenida la tripsinización, las células se tiñeron inmediatamente con azul tripán al 0,5% y se determinó el número de células vivas y muertas en una cámara de Neubauer. Los resultados se presentan comparando los porcentajes de células vivas respecto al control. Se realizó un mínimo de cinco experimentos independientes para cada uno de los análisis.
Preparación de extractos celulares y análisis por transferencia Western
Una vez realizados los tratamientos, las células se lavaron, se recogieron y se solubilizaron en tampón Tris-HCl 10 mM pH 7,4, Triton X-100 al 1% y PMSF 0,1 mM. Posteriormente se centrifugaron durante 20 min a 10.000 G y el contenido proteínico de los sobrenadantes se determinó mediante el método del ácido bicinconínico. Para la determinación de la fosforilación de SAPK/JNK, ERK1/2 y p-38 MAPK, los cardiocitos se lisaron en 50 μl del tampón Tris-HCl 10 mM pH 7,4, PMSF 1 mM, Igepal CA-630 al 1% y cóctel inhibidor de fosfatasas al 1%.
La activación de las caspasas 3, 8 y 9 y la fosforilación de SAPK/JNK, ERK1/2 y p-38 MAPK se determinaron mediante análisis por transferencia Western. Para ello, se separaron 20 μg de cada extracto proteínico mediante electroforesis SDS-PAGE y se transfirieron a membranas de PVDF (Millipore, Bedford, Massachusetts, Estados Unidos). Éstas se bloquearon con leche desnatada al 5% en PBS y se incubaron durante toda la noche a 4 °C y en tampón de unión (PBS, Tween 20 al 0,1%) con el anticuerpo primario correspondiente para cada proteína (1:1.000). Al día siguiente, las membranas se lavaron y se incubaron durante 60 min a temperatura ambiente con el anticuerpo secundario marcado con peroxidasa (1:5.000). Tras repetir los lavados, la unión del anticuerpo se detectó por quimioluminiscencia con ECL (GE Healthcare Biosciences, Países Bajos). Se realizó un mínimo de cuatro experimentos independientes para cada una de las proteínas analizadas. El análisis cuantitativo se realizó con el programa Gel Pro Analyzer 3.1 (Sigma).
Determinación de fragmentación del ADN
Para la extracción del ADN, las células se lisaron en tris-HCl 50 mM pH 7,4, EDTA 20 mM e Igepal CA-630 y se incubaron, primero en SDS al 2% y RNasa 5 μg/μl durante 2 h a 56 °C y después en proteinasa K 2,5 μg/μl durante 2 h a 37 °C. Después el ADN se precipitó en etanol y se resuspendió finalmente en agua destilada. La fragmentación del ADN se analizó mediante separación electroforética en geles de agarosa al 2% y posterior tinción con solución de bromuro de etidio 0,2 μg/ml.
Análisis estadístico
Los resultados de este trabajo se muestran como media ± desviación estándar para un número n de experimentos independientes. Las diferencias entre grupos se analizaron mediante la prueba no paramétrica de la U de Mann-Whitney. Se consideraron significativos los valores de p < 0,05.
RESULTADOS
Apoptosis y testosterona
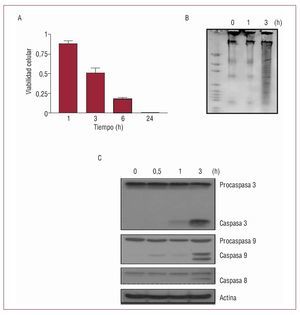
La inducción de estrés hiperosmótico en las células H9c2 mediante tratamiento con el soluto no permeable sorbitol (0,6 M) dio lugar a una disminución de la viabilidad celular rápida y dependiente del tiempo, un incremento de la fragmentación del ADN y una activación de las caspasas proteolíticas 3, 8 y 9 (fig. 1). El tiempo de tratamiento con sorbitol seleccionado para la inducción de apoptosis en análisis posteriores fue de 3 h, tiempo en el que todos los efectos anteriores fueron significativos.

Fig. 1. El estrés hiperosmótico induce una apoptosis rápida y dependiente del tiempo en las células H9c2. Las células se incubaron durante diferentes tiempos con sorbitol 0,6 M en medio de cultivo libre de suero y antibióticos. A: viabilidad celular; los valores se representan como la media ± desviación estándar (cinco experimentos independientes). B: activación de la fragmentación del ADN. C: activación de las caspasas 3, 8 y 9, analizadas mediante transferencia Western, como respuesta a la presencia de sorbitol.
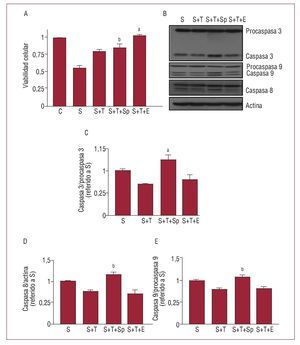
Los efectos del tratamiento combinado de sorbitol con testosterona y/o flutamida en la viabilidad celular y la activación de las caspasas se muestran en la figura 2. Destaca que la administración de testosterona (T) o flutamida (F) en ausencia de sorbitol no tuvo efecto alguno. Al comparar con sorbitol sólo (S), el tratamiento combinado con testosterona se asoció con un incremento en la viabilidad celular (S+T frente a S, p < 0,001). Además, al añadir flutamida, antagonista del receptor de andrógenos, no se alteró este efecto protector de la testosterona (S+T+F frente a S+T, sin significación). Del mismo modo, el tratamiento combinado con testosterona redujo la activación inducida por sorbitol en todas las caspasas analizadas (S+T frente a S, p < 0,01) y la adición de flutamida incrementó dicha inhibición (S+T+F frente a S+T, p < 0,05).

Fig. 2. Efectos de la testosterona en la apoptosis inducida por sorbitol. A: viabilidad celular. B: transferencia Western de la activación de las caspasas. C-E: análisis densitométrico calculado como caspasa activa/procaspasa total para las caspasas 3 y 9, y como caspasa activa/actina para la caspasa 8. Los resultados se muestran como media ± desviación estándar (seis experimentos independientes). C: control; F: flutamida; S: sorbitol; T: testosterona. ap < 0,001 respecto a sorbitol. bp < 0,001 respecto a sorbitol + testosterona. cp < 0,01 respecto a sorbitol. dp < 0,05 respecto a sorbitol + testosterona.
La figura 3 muestra el análisis de las MAPK implicadas en la apoptosis inducida por sorbitol. Así, el tratamiento con sorbitol (1 h) indujo la fosforilación de SAPK/JNK y disminuyó la fosforilación de ERK1/2, sin mediar ningún efecto en la p38 MAPK (datos no mostrados). La administración simultánea de testosterona revirtió significativamente los efectos del sorbitol, disminuyó la activación de SAPK/JNK e incrementó la activación de ERK1/2. Estos efectos de testosterona tampoco fueron alterados por la presencia de flutamida.

Fig. 3. Efectos de la testosterona en las cascadas de señalización de las MAPK. Transferencia Western y análisis densitométrico de la activación de (A) JNK y (B) ERK1/2. Tras 1 h de tratamiento, la activación de las cinasas se calculó como proteína fosforilada / proteína total. Los resultados se muestran como media ± desviación estándar (cuatro experimentos independientes). C: control; F: flutamida; S: sorbitol; T: testosterona. ap < 0,001 respecto a sorbitol. bp < 0,01 respecto a sorbitol.
Testosterona, espironolactona y eplerenona
La figura 4 muestra los efectos de espironolactona y eplerenona en la disminución de la apoptosis mediada por testosterona. La administración de espironolactona o eplerenona no tuvo efecto propio en la viabilidad celular o la activación de las caspasas en las células H9c2 ni causó cambio alguno en la apoptosis inducida por el sorbitol (datos no mostrados). Sin embargo, la espironolactona y la eplerenona tuvieron efectos opuestos en el papel protector de la testosterona. Así, el pretratamiento con espironolactona bloqueó los efectos de testosterona, disminuyó la viabilidad celular (S+T+Sp frente a S+T, p < 0,01) e incrementó la activación de las caspasas 3, 8 y 9 (S+T+Sp frente a S+T, p < 0,01). Por el contrario, el pretratamiento con eplerenona incrementó la viabilidad celular (S+T+Ep frente a S+T, p < 0,001) y no tuvo efecto en la disminución de la activación de las caspasas inducida por testosterona (S+T+Ep frente a S+T, no significativo).

Fig. 4. Efectos de la espironolactona y la eplerenona en la viabilidad celular y la activación de las caspasas. A: viabilidad celular. B: transferencia Western de la actividad de las caspasas. C-E: análisis densitométrico para cada caspasa. Los resultados se muestran como media ± desviación estándar (cinco experimentos independientes). C: control; E: eplerenona; F: flutamida; S: sorbitol; Sp: espironolactona; T: testosterona. ap < 0,001 respecto a sorbitol + testosterona. bp < 0,01 respecto a sorbitol + testosterona.
DISCUSIÓN
Los principales hallazgos de este estudio son: a) la testosterona reduce la apoptosis inducida por estrés hiperosmótico en la línea de cardiomiocitos H9c2; b) esta acción no está mediada por el receptor de andrógenos e implica a las cascadas de señalización de SAPK/JNK y ERK1/2, y c) espironolactona, pero no por eplerenona, contrarresta este efecto protector de testosterona, lo que indica efectos diferentes de uno y otro fármaco.
En este estudio, hemos usado el estrés hiperosmótico como inductor de la apoptosis, por tratarse de uno de los principales mecanismos de daño tisular en estados patológicos como la isquemia, el choque séptico y la acidosis14. Además es un modelo de inducción de apoptosis más rápido y potente que otros modelos clásicos como la hipoxia, la doxorubicina o la angiotensina II15, lo que nos permitió abordar el estudio de los posibles efectos no genómicos de la testosterona en la apoptosis inducida. Éste es el primer trabajo que demuestra que la testosterona, mediante una ruta rápida no genómica, protege a los cardioblastos H9c2 de la apoptosis inducida por estrés hiperosmótico. Esta hipótesis se confirma por el hecho de que la flutamida, un bloqueador del receptor de andrógenos, no bloqueó el efecto protector de la testosterona. Sin embargo, aún falta determinar los mecanismos implicados en este efecto beneficioso de la testosterona.
Está bien establecido que la ruta clásica de acción de la testosterona implica su unión al receptor de andrógenos y la translocación del complejo testosterona-receptor al núcleo, que activa finalmente la síntesis de proteínas16. Recientemente, se ha demostrado en cardiocitos17,18 y otros sistemas celulares6 una acción rápida y no genómica de la testosterona independiente de su receptor de andrógenos. Así, mediante su unión a un receptor de membrana acoplado a proteína G (GPCR), la testosterona es capaz de activar la cascada PLC/I3P e inducir un rápido incremento intracelular del calcio en cultivo primario de cardiocitos de rata18. En células del músculo esquelético, vía unión a un GPCR y por incremento del calcio intracelular, la testosterona es capaz de activar la ruta de señalización MEK/ERK y mediar un efecto antiapoptótico6 similar al obtenido en nuestros resultados. Por otro lado, se ha demostrado que la testosterona es un fuerte bloqueador de los canales de calcio tipo L19, los cuales participan en la activación de la apoptosis en numerosos sistemas celulares, entre ellos los cardiocitos20.
Nuestros resultados demuestran que las cascadas ERK1/2 y SAPK/JNK están implicadas en la inhibición de la apoptosis mediada por la testosterona. Sin embargo, son necesarios nuevos experimentos para conocer si este efecto protector se debe al bloqueo de los canales de calcio tipo L o al incremento del calcio intracelular a través de su unión a un GPCR. Cabe discutir que otros grupos han demostrado previamente un efecto proapoptótico de la testosterona en células cardiacas21, pero en esos estudios el efecto adverso de la testosterona está mediado por largas exposiciones (≥ 20 h) a la hormona, lo que indica una vía genómica de acuerdo con la ruta clásica de la testosterona a través de su unión al receptor de andrógenos, diferente del efecto rápido de la testosterona descrito en nuestro estudio. Del mismo modo, se sabe que los andrógenos median efectos protectores contra el catabolismo inducido por los glucocorticoides22. Los receptores de glucocorticoides, al igual que el receptor de andrógenos, son receptores citoplásmicos que, al unirse a su ligando, se translocan al núcleo donde activan finalmente la síntesis de proteínas. Se trata de un mecanismo de acción lento desde la unión del ligando hasta la expresión de proteínas. Si el efecto antiapoptótico de la testosterona se debiera a su capacidad de bloquear un efecto apoptótico mediado por los glucocorticoides, tal efecto no se vería hasta momentos muy posteriores (24 h) a los descritos en nuestros resultados. Este aspecto nos permite descartar el receptor de glucocorticoides como posible diana que explique los efectos rápidos no genómicos descritos en este estudio.
La espironolactona y la eplerenona son dos fármacos usados habitualmente en el tratamiento de la insuficiencia cardiaca23, pero tienen diferencias estructurales significativas. La espironolactona es un antimineralocorticoide al que se atribuyen efectos progestágenos y antiandrogénicos11, mientras que la eplerenona es un derivado de la espironolactona diseñado para incrementar su selectividad por el receptor de mineralocorticoides al que se han minimizado esos efectos11,12. En nuestro estudio, la espironolactona redujo los efectos protectores de la testosterona, mientras que la eplerenona no tuvo ningún efecto de bloqueo. Estas acciones diferentes de espironolactona y eplerenona no pueden atribuirse a sus diferentes capacidades por la unión al receptor de andrógenos, ya que el efecto protector de la testosterona fue independiente de dicho receptor. Las diferencias podrían deberse a los efectos progestágenos de la espironolactona ausentes en la eplerenona. En este sentido, se sabe que la progesterona media efectos no genómicos en diferentes tipos de tejidos24, como la modulación de los flujos de calcio25, de las concentraciones de adenosinmonofosfato cíclico (AMPc)26 y la señalización de MAPK27. Por lo tanto, al presentar elementos estructurales de la molécula de progesterona y efectos progestágenos11, la espironolactona contrarrestaría la acción antiapoptótica de la testosterona. La eplerenona, cuyos efectos progestágenos han sido eliminados, no contrarrestaría el efecto protector de la testosterona. Actualmente, no hay estudios que demuestren esta hipótesis, tan sólo algunos trabajos en los que la espironolactona indujo rápidos incrementos de calcio intracelular en el miocardio28, pero no profundizan en los mecanismos implicados. Por todo esto, nuestros resultados señalan la necesidad de nuevos estudios que aborden los mecanismos inducidos por la espironolactona, pero no por la eplerenona, implicados en el bloqueo de los efectos beneficiosos de la testosterona.
Implicaciones clínicas
Los hallazgos de este estudio podrían tener importantes implicaciones clínicas en cuanto a que la apoptosis es un mecanismo de muerte celular en la insuficiencia cardiaca, en la que además se ha demostrado que el déficit de testosterona se asocia a un peor pronóstico2-4. Nuestros resultados indican que el déficit anabólico presente en estados avanzados de la insuficiencia cardiaca podría ser un factor determinante del mayor grado de apoptosis descrito en la enfermedad y, por lo tanto, de una peor evolución clínica. En la actualidad, el tratamiento con testosterona en la insuficiencia cardiaca representa una atractiva opción terapéutica en evaluación, que ha demostrado mejorar los síntomas, la capacidad de ejercicio y la calidad de vida en algunos ensayos con pequeño número de pacientes2,29. Sin embargo, su efecto en la evolución de la enfermedad no se ha evaluado todavía29.
Por otro lado, ya se sabe que la espironolactona y la eplerenona son dos fármacos de uso habitual en la insuficiencia cardiaca avanzada debido a sus efectos beneficiosos como bloqueadores del receptor de aldosterona. Las diferencias entre ambos fármacos, en cuanto a menor tasa de efectos secundarios de la eplerenona por su escasa acción antiandrogénica, son bien conocidas. Sin embargo, hasta ahora no se han descrito diferencias en sus acciones en el corazón y tampoco existen estudios comparativos directos entre ambos fármacos. Nuestro estudio es el primero en mostrar en el cardiocito las diferencias de selectividad entre espironolactona y eplerenona sobre la acción de la testosterona. Los hallazgos indican que los beneficios de la espironolactona en el cardiomiocito podrían estar disminuidos porque se bloquea el efecto protector de la testosterona. Por el contrario, la eplerenona podría tener una ventaja respecto a la espironolactona, ya que no disminuyó la acción antiapoptótica de la testosterona. Sin embargo, se necesitan nuevos estudios básicos y clínicos que evalúen esta posibilidad en modelos in vivo.
Limitaciones
Los cardiocitos empleados en este estudio fueron la línea celular embrionaria de mioblastos cardiacos H9c2, los cuales podrían tener diferencias fenotípicas respecto a los cardiocitos adultos. La espironolactona y la eplerenona se usaron a concentraciones que, si bien son similares a las de otros estudios in vitro, no son equiparables a las habitualmente usadas en la práctica clínica y tampoco se realizó un estudio de dosis-respuesta. Por todo ello, los resultados presentados en este trabajo deberían confirmarse en futuros estudios que se desarrollen en modelos in vivo, usando concentraciones equiparables a las de la práctica clínica. Estos modelos de insuficiencia cardiaca deberán ser preferiblemente crónicos y tomar en consideración las posibles interacciones con otros tipos celulares, como los fibroblastos, así como los fenómenos de fibrosis y remodelado, no evaluados por este trabajo in vitro.
CONCLUSIONES
Este estudio demuestra que el tratamiento con testosterona protege la línea celular embrionaria cardiaca H9c2 de la apoptosis inducida por estrés hiperosmótico, a través de mecanismos no genómicos e independientes del receptor de andrógenos, en el que al menos están implicadas SAPK/JNK y ERK1/2. Además, este efecto beneficioso de la testosterona es bloqueado por espironolactona pero no por eplerenona, lo que indica acciones diferentes de uno y otro fármaco y un posible beneficio adicional de la eplerenona respecto a la espironolactona.
AGRADECIMIENTOS
Al Prof. F. Fernández-Belda (Facultad de Veterinaria, Murcia, España) por la cesión desinteresada de la línea celular H9c2.
ABREVIATURAS
GPCR: receptor de membrana acoplado a proteína G.
MAPK: proteincinasa activada por mitógenos.
Full English text available from: www.revespcardiol.org
VÉASE EDITORIAL EN PÁGS. 760-2
Este trabajo fue financiado parcialmente por una beca de la Fundación Séneca (Agencia de Ciencia y Tecnología de la Región de Murcia) (05822/ PPC/07), la red nacional de investigación en insuficiencia cardiaca REDINSCOR (Ministerio de Sanidad y Consumo) (RD06/0003/0013) y una beca de Pfizer Inc. (New York, Estados Unidos).
Correspondencia: Dr. D.A. Pascual Figal.
Unidad de Insuficiencia Cardiaca. Servicio de Cardiología. Hospital Universitario Virgen de la Arrixaca.
Ctra. Madrid-Cartagena, s/n. 30120 El Palmar. Murcia. España.
Correo electrónico: dapascual@servicam.com
Recibido el 21 de noviembre de 2009.
Aceptado para su publicación el 25 de enero de 2010.