El edoxabán es un inhibidor potente, directo, selectivo y reversible del factor Xa de la coagulación. Por su mecanismo de acción, prolonga los tiempos de protrombina y de tromboplastina parcial activada e inhibe la actividad anti-factor Xa. En modelos in vitro e in vivo, el edoxabán es más efectivo como antitrombótico que el fondaparinux. Por vía oral, alcanza rápidamente (1-2 h) concentraciones plasmáticas máximas y su efecto máximo en diversos biomarcadores antitrombóticos (tiempos de protrombina y de tromboplastina parcial activada, actividad anti-factor Xa). El edoxabán (15-150 mg una vez al día) presenta un perfil farmacocinético lineal. Se absorbe bien por vía oral (biodisponibilidad del 62%) pero, a diferencia de otros inhibidores del factor Xa, se une poco a proteínas plasmáticas y no se biotransforma a través del citocromo P3A4, se elimina por vía biliar y renal y tiene una semivida de 10-14 h, lo que permite su administración una vez al día. La exposición al edoxabán es independiente de la edad, el sexo o la raza, pero aumenta al disminuir el peso y la función renal del paciente.
Palabras clave
aclaramiento de creatinina
citocromo P450
transportadores de aniones orgánicos
tiempo de trombina
tiempo de tromboplastina parcial activada
factor X activado de la coagulación
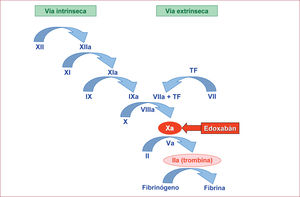
El factor X activado (Xa) de la coagulación es una serina proteasa que se encuentra en el punto en que convergen las vías intrínseca y extrínseca de la coagulación (figura 1). El factor Xa se une al factor Va, Ca2+ y los fosfolípidos en la superficie de las plaquetas y forma el complejo protrombinasa, que activa la conversión del factor II (protrombina) en factor Ila (trombina)1. De hecho, una molécula de factor Xa puede generar unas 10.000 moléculas de trombina2. Esto convierte al factor Xa en una importante diana terapéutica.

El edoxabán produce una inhibición directa (no requiere de antitrombina III como cofactor), selectiva, competitiva, reversible y dependiente de la dosis (constante de inhibición [Ki], 0,4 nmol/l) del factor Xa unido al trombo (Ki, 0,561 nm) o al complejo de la protrombinasa en la superficie plaquetaria (Ki, 2,98 nm). Su afinidad por el factor Xa es unas 10.000 veces mayor que por otras serinaproteasas (Cl50 > 20 μmol/l), como trombina, plasmina, urocinasa, proteína C activada o los factores de la coagulación Vlla y IXa3. Como consecuencia, el edoxabán reduce de manera dependiente de la dosis la generación de trombina (GT) y la formación del trombo3–6. La Agencia Europea del Medicamento (EMA) ha aprobado el edoxabán para: a) la prevención del ictus y la embolia sistémica en pacientes adultos con fibrilación auricular no valvular y uno o más factores de riesgo, tales como insuficiencia cardiaca congestiva, hipertensión, edad ≥ 75 años, diabetes mellitus, ictus o accidente isquémico transitorio previos, y b) el tratamiento de la trombosis venosa profunda (TVP) y la embolia pulmonar, y prevención de las recurrencias de estas en adultos. En este artículo se analizan las propiedades farmacodinámicas y farmacocinéticas del edoxabán, así como la correlación entre ambas.
Propiedades farmacodinámicasEstudios in vitroEn plasma humano, el edoxabán inhibe la GT y es 3 veces más potente que el fondaparinux; a la vez, a concentraciones de 0,256 y 0,508 pm duplica los tiempos de trombina (PT) y de tromboplastina parcial activada (TTPa); también prolonga el INR (cociente internacional normalizado), pero no modifica el tiempo de ecarina3,7. El edoxabán no modifica la agregación inducida por colágeno, adenosina difosfato (ADP), U46619 (un análogo de la prostaglandina H2/tromboxano A2) o trombina en plaquetas humanas, pero sí antagoniza la agregación inducida por el factor tisular y el factor Xa unido al coágulo de modo más potente que el fondaparinux, un inhibidor indirecto de Xa3,8.
El edoxabán y el fondaparinux, un inhibidor indirecto del Xa, se han comparado en un modelo de trombosis ex vivo utilizando una cámara de perfusión en la que se simulan las condiciones de alta y baja velocidad de cizallamiento existentes en, respectivamente, las venas y arterias estenosadas. En este modelo, el cociente de la dosis eficaz 50% (DE50) en condiciones de alto (1,13 mg/kg/h) y bajo cizallamiento (0,63 mg/kg/h) era de 1,19, mientras que para fondaparinux era > 669.
El edoxabán (3 mg/kg/h) también inhibía la formación de fibrina en condiciones de baja y alta velocidad de cizallamiento, y reducía el peso del trombo en un 88% en condiciones de baja velocidad de cizallamiento y en un 70% en condiciones de alta velocidad de cizallamiento. Sin embargo, el fondaparinux inhibía en un 84% la formación del trombo venoso, pero solo producía una ligera reducción de la formación de fibrina y del peso del trombo en condiciones de alta velocidad de cizallamiento9. En otro estudio, un grupo de voluntarios sanos recibió 60 mg de edoxabán, y sus efectos antitromboticos se analizaron ex vivo estudiando la formación de trombos en condiciones que reproducían los flujos venoso y arterial10. En condiciones de flujo venoso, el tamaño del trombo disminuía en un 28 y un 21% al cabo de 1,5 y 5 h respectivamente (p > 0,05), mientras que en condiciones que semejan el flujo arterial la reducción del trombo era del 26 y el 17% (p < 0,05). La generación de trombina también se reducía en un 28 y un 10% respectivamente10.
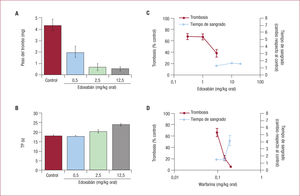
Estudios in vivoEn modelos animalesEl edoxabán inhibe de manera dependiente de la dosis la formación de trombos venosos (ricos en fibrina, pobres en plaquetas) inducidos en ratas tras la ligadura de la vena cava inferior justo por debajo de la vena renal o la introducción de un hilo de platino en la vena cava inferior (figura 2A), o en conejos tras la ligadura de la vena auricular, así como la formación de trombos arteriales inducidos por el cloruro férrico (FeCl3) en la arteria carótida3. Las dosis de edoxabán que reducían la formación de trombos en un 50% (DE50) en modelos de trombosis venosa y arterial, eran 0,076 y 0,093 mg/kg/h, respectivamente, mientras que la DE50 de fondaparinux en el modelo de trombosis arterial (> 10 mg/kg/h) era muy superior a la observada en el modelo de trombosis venosa (0,021 mg/kg/h)9. El edoxabán, además, prolongaba el TP (figura 2B), inhibía la actividad del factor Xa exógeno y prolongaba significativamente (1,9 veces) el tiempo de sangrado en la cola de rata respecto al grupo control.
. Curvas dosis-respuesta del efecto antitrombótico y del tiempo de sangrado en ratas tratadas con edoxabán (C) y warfarina (D) (reproducido con permiso de Morishima et al11). TP: tiempo de protrombina.")
A: efecto antitrombótico del edoxabán en un modelo murino de trombosis producida por estasis venosa. B: efecto en el tiempo de protrombina (reproducido con permiso de Furugohri et al3). Curvas dosis-respuesta del efecto antitrombótico y del tiempo de sangrado en ratas tratadas con edoxabán (C) y warfarina (D) (reproducido con permiso de Morishima et al11). TP: tiempo de protrombina.
En un estudio comparativo de los efectos antitrombóticos y sobre el tiempo de hemorragia de edoxabán, warfarina y enoxaparina realizado en ratas (figuras 2C y D), el índice terapéutico era > 10,5 con edoxabán y 1,3 y 3,4, respectivamente, con warfarina y enoxaparina, lo que indica que el edoxabán presenta mayor margen de seguridad11.
También se han analizado los efectos combinados de la administración oral de edoxabán, ácido acetilsalicílico y clopidogrel a ratas en ayunas, 0,5-2 h antes de la inducción de un trombo8. La combinación de dosis submáximas de edoxabán (1 mg/kg) y ácido acetilsalicílico (50 mg/kg), o de edoxabán y clopidogrel (10 mg/kg) producía efectos antitrombóticos aditivos. El edoxabán y el ácido acetilsalicílico en monoterapia no modificaban el tiempo de sangrado, pero su combinación prolongaba casi 2 veces este parámetro. El clopidogrel también prolonga el tiempo de sangrado 2,1 veces, pero la adición de edoxabán no modificaba los cambios producidos del clopidogrel.
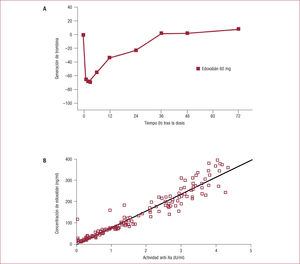
Ensayos clínicosEn voluntarios sanos, el edoxabán (60 mg/día) inhibe la GT durante 24 h12, lo que avala la administración del fármaco una vez al día (figura 3A). Además, el edoxabán prolonga de manera dependiente de la dosis el TTPa, la actividad anti-Xa (figura 3B) y el INR12,13. Sin embargo, los cambios en TP o TTPa son pequeños, tienen gran variabilidad y no son útiles para controlar el efecto anticoagulante de edoxabán14. La determinación cromogénica de la actividad antiXa calibrada para edoxabán es la prueba ideal, aunque no se dispone de valores que definan claramente el riesgo de sangrado o de trombosis15–17. Debido a la semivida del edoxabán, su efecto en las pruebas de coagulación disminuye progresivamente a partir de las primeras horas de su administración, por lo que el resultado de cualquier prueba de coagulación se debe interpretar teniendo en cuenta el tiempo transcurrido entre la ingesta del fármaco y el momento en que se toma la muestra de sangre. Cuando se administra edoxabán 24 h tras la administración de warfarina, se observa un rápido aumento en el TP (máximo al cabo de 2 h), que vuelve a los valores control al cabo de 12 h4–6, y un aumento en el TTPa (de 50,8 hasta 67,4 s al cabo de 1 h). Sin embargo, la warfarina no modificaba la actividad anti-Xa del edoxabán18.
. B:relación entre las concentraciones plasmáticas de edoxabán y la actividad anti-Xa (reproducido con permiso de Mendell et al18).")
A: cambios en la generación de trombina tras la administración de una dosis de 60 mg de edoxabán a voluntarios sanos (reproducido con permiso de Zahir et al12). B:relación entre las concentraciones plasmáticas de edoxabán y la actividad anti-Xa (reproducido con permiso de Mendell et al18).
El edoxabán (30-120 mg/día) produce una rápida disminución dependiente de la dosis de los marcadores de la coagulación (fragmentos 1+2 de la protrombina y complejo trombina-antitrombina) y de la activación plaquetaria (tromboglobulina beta), que alcanzan su máximo al cabo de 1,5 h y persisten alterados durante 24 h, y su efecto es superior al del fondaparinux (2,5 mg)19. En los pacientes con TVP de los estudios STARS E-3 y STARS JV, la reducción de la concentración plasmática de dímero D, fragmentos 1+2 de la protrombina y complejos de monómeros de fibrina soluble producida por el edoxabán (30 mg/día) era significativamente mayor que la producida por la enoxaparina al cabo de 7-14 días de tratamiento20.
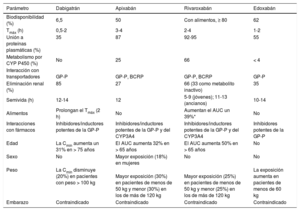
Características farmacocinéticasEn voluntarios sanos, el edoxabán presenta una farmacocinética lineal tras la administración de dosis únicas (15-150 mg/día) o repetidas (60-120 mg/día)21. Por vía oral se absorbe bien (biodisponibilidad, 62%)6,22 y rápidamente, pues alcanza concentraciones plasmáticas máximas (Cmáx, 303 ± 88 ng/ml tras la administración de 60 mg) al cabo de 1-2 h y valores estables al cabo de 3 días6,21–24. Las características farmacocinéticas no se modifican en presencia de alimentos (incluso de una dieta grasa), en pacientes con aclorhidria o cuando se administra con inhibidores de la bomba de protones23,25,26. La tabla compara las propiedades farmacocinéticas de los nuevos anticoagulantes orales directos.
Propiedades farmacocinéticas de los anticoagulantes orales.
| Parámetro | Dabigatrán | Apixabán | Rivaroxabán | Edoxabán |
|---|---|---|---|---|
| Biodisponibilidad (%) | 6,5 | 50 | Con alimentos, ≥ 80 | 62 |
| Tmáx (h) | 0,5-2 | 3-4 | 2-4 | 1-2 |
| Unión a proteínas plasmáticas (%) | 35 | 87 | 92-95 | 55 |
| Metabolismo por CYP P450 (%) | No | 25 | 66 | < 4 |
| Interacción con transportadores | GP-P | GP-P, BCRP | GP-P, BCRP | GP-P |
| Eliminación renal (%) | 85 | 27 | 66 (33 como metabolito inactivo) | 35 |
| Semivida (h) | 12-14 | 12 | 5-9 (jóvenes); 11-13 (ancianos) | 10-14 |
| Alimentos | Prolongan el Tmáx (2 h) | No | Aumentan el AUC un 39%* | No |
| Interacciones con fármacos | Inhibidores/inductores potentes de la GP-P | Inhibidores/inductores potentes de la GP-P y del CYP3A4 | Inhibidores/inductores potentes de la GP-P y del CYP3A4 | Inhibidores potentes de la GP-P |
| Edad | La Cmín aumenta un 31% en > 75 años | El AUC aumenta 32% en > 65 años | El AUC aumenta 50% en > 65 años | No |
| Sexo | No | Mayor exposición (18%) en mujeres | No | No |
| Peso | La Cmín disminuye (20%) en pacientes con peso > 100 kg | Mayor exposición (30%) en pacientes de menos de 50 kg y menor (30%) en los de más de 120 kg | Mayor exposición (25%) en pacientes de menos de 50 kg y menor (25%) en los de más de 120 kg | La exposición aumenta en pacientes de menos de 60 kg |
| Embarazo | Contraindicado | Contraindicado | Contraindicado | Contraindicado |
AUC: área bajo la curva de las concentraciones plasmáticas; BCRP (ABCG2): proteína de resistencia de cáncer de mama; CmIn: concentraciones plasmáticas al final del intervalo entre dosis; GP-P: glucoproteína P; Tmáx: tiempo hasta alcanzar las concentraciones plasmáticas máximas.
El edoxabán se une a las proteínas plasmáticas en un 55%, porcentaje muy inferior a los del apixabán (87%) y rivaroxabán (~90%), y su volumen de distribución es de 1,5 l/kg4–6; se observa que, tras la administración repetida una vez al día, no se produce una acumulación relevante del fármaco (1,14)21. Una pequeña proporción del fármaco administrado (< 10%) sufre procesos de hidrolisis, conjugación y oxidación (CYP3A4/5) en el hígado, por lo que los fármacos inductores/ inhibidores de estos procesos apenas si interaccionan con el edoxabán. La hidrolisis se realiza por la carboxilesterasa 1 hepática y conduce a la formación de un metabolito (M-4) activo que alcanza menos del 10% de la exposición de edoxabán6,27–29; la exposición de los restantes metabolitos es menor del 5%. Por la baja concentración plasmática que alcanza y su alta unión a proteínas plasmáticas (80%), no es de esperar que el M-4 contribuya de manera importante a los efectos del edoxabán en individuos con un aclaramiento de creatinina (AclCr) > 60 ml/min30. El edoxabán se elimina sin biotransformar en la orina en un 50% (aclaramiento renal a 11 l/h) y el otro 50% por vía biliar/fecal, y su semivida de eliminación es de 10-14 h21,22,25,29.
In vitro, el edoxabán es un sustrato de la glucoproteína P (GP-P), una bomba que facilita la salida del fármaco y sus metabolitos de la célula en distintos tejidos (enterocitos, hepatocitos, células renales), reduce su absorción y acelera su eliminación intestinal, biliar o renal31. Por lo tanto, los inhibidores potentes de la GP-P (amiodarona, drone-darona, quinidina, verapamilo) aumentan la exposición al edoxabán (expresada por las Cmáx y el área bajo la curva [AUC] de las concentraciones plasmáticas), aunque no llegan a duplicarla. Los inhibidores potentes de la GP-P y del CYP3A4/5 (cetoconazol, eritromicina) no producen un aumento de la exposición del edoxabán superior al producido por fármacos que bloquean la GP-P e inhiben ligeramente el CYP3A4/5, lo que confirma que el metabolismo del edoxabán a través del CYP3A4/5 es poco importante. Sin embargo, el edoxabán no inhibe/induce diversas isoformas del citocromo P450 (CYP1A2, 2A6, 2B6, 2C8/9, 2C19, 2D6, 2E1 o 3A4), los transportadores de aniones orgánicos OAT1 y OAT3 o de cationes orgánicos OCT1 y OCT2 o los polipéptidos transportadores de aniones orgánicos (OATP1B1 y OATP1B3)6,25,32.
Poblaciones especialesLas propiedades farmacocinéticas del edoxabán son independientes del sexo33 y la raza34. En pacientes ancianos aumenta el AUC y disminuye el aclaramiento de edoxabán, posiblemente como consecuencia de la disminución de la función renal con la edad. El estudio ENGAGE AF-TIMI35 incluyó a 5.182 pacientes de edad ≥ 65 años y 2.838 de edad ≥ 75 años, y en el estudio Hokusai-TEV36, 1.334 pacientes (32%) tenían 65 o más años y 560 (14%), 75 o más. En ambos estudios, las incidencias de reacciones adversas en pacientes mayores y menores de 65 años fueron similares. En pacientes con bajo peso corporal (< 55 kg), la exposición total a edoxabán aumenta un 13% respecto a los pacientes de más peso corporal (84 kg). Por ello, se recomienda utilizar la dosis de 30 mg/día para los pacientes cuyo peso corporal sea < 70 kg35. Igualmente, en pacientes japoneses con fibrilación auricular, la Cmín de edoxabán era 1,8 veces mayor en los pacientes de peso < 60 kg que en los de más de 60 kg37. No se ha estudiado la seguridad y la eficacia del edoxabán en la población pediátrica.
No hay diferencias en la farmacocinética del edoxabán en pacientes con insuficiencia hepática leve o moderada (Child-Pugh A o B), pero no hay experiencia en pacientes con insuficiencia hepática grave que presenten defectos intrínsecos de la coagulación4–6.
La seguridad y la eficacia del edoxabán no se han analizado en mujeres embarazadas y se desconoce si se elimina por la leche materna, por lo que solo se debe utilizar cuando el beneficio potencial así lo justifique (categoría C)4–6,38.
Insuficiencia renalEl 50% del edoxabán se elimina por vía renal y su concentración plasmática aumenta en pacientes con insuficiencia renal. En un estudio abierto de 8 semanas de duración realizado en pacientes con fibrilación auricular e insuficiencia renal (IR) grave, el edoxabán (15 mg/día durante 8 semanas) produjo concentraciones plasmáticas, cambios en el TP y tasa de sangrado similares a los observados tras la administración de 30 o 60 mg/día en pacientes con IR moderada o función renal normal39. En pacientes con TVP sometidos a cirugía ortopédica de extremidades inferiores, los que tenían IR grave recibieron 15 mg de edoxabán y los que la tenían leve, 30 mg20. Al cabo de 7 días de tratamiento, las concentraciones plasmáticas de edoxabán y las incidencias de reacciones adversas y de sangrado eran similares en ambos grupos. Finalmente, en otro estudio realizado en pacientes con AclCr > 50-< 80, 30-50 y < 30 ml/min o en diálisis peritoneal, los valores plasmáticos aumentaban, respectivamente, en un 32, un 74, un 72 y un 93% frente a los pacientes con AclCr ≥ 80 ml/min40. Estos resultados explican por qué en el estudio ENGAGE AF-TIMI 48 la dosis de edoxabán se reducía en un 50% en los pacientes con AclCr 30-50 ml/min, y no se recomienda su uso en pacientes con AclCr < 15 ml/min.
En pacientes con enfermedad renal terminal, la exposición al edoxabán disminuía solo en un 7% tras 4 h de diálisis, por lo que no es necesario reajustar la dosis4–6,21,22,40,41.
Relación entre las propiedades farmacodinámicas y farmacocinéticasTras la administración de edoxabán (10-150 mg) a voluntarios sanos, se observa una correlación lineal entre las Cmáx y el curso temporal de los marcadores que indican su acción antitrombótica (actividad anti-Xa, TP, TTPa y GT). El efecto máximo en estos marcadores se alcanza al cabo de 1-3 h (coincidiendo con las Cmáx) y persisten elevados durante unas 24 h4–6,12,21,41,42. Se ha observado una relación similar en pacientes con TVP o fibrilación auricular4–6,25.
En un estudio de fase II, se observó que las concentraciones plasmáticas mínimas en estado estacionario (Cmín,ss) y Cmáx, el AUC entre las 0 y las 24 h en estado estacionario (AUCss) y la incidencia de hemorragias, mayores o no, era significativamente mayor con edoxabán que con warfarina si aquel se administraba en dosis de 30 o 60 mg 2 veces al día, pero estas diferencias desaparecían cuando el edoxabán se administraba una vez al día43.
Finalmente, el modelado de los resultados de los estudios de fase I y II indican que las dosis de 30 y 60 mg una vez al día son las más adecuadas para realizar los estudios de fase III con TVP o fibrilación auricular y que es necesario reducir en un 50% la dosis para los pacientes con peso corporal < 60 kg, IR moderada (AclCr 30-50 ml/ min) o que requieran la administración de inhibidores potentes de la GP-P36,44,45
ConclusionesEl edoxabán es un inhibidor directo, selectivo y reversible del factor Xa; hay correlación entre sus propiedades farmacocinéticas y farmacodinámicas, que alcanzan su máximo rápidamente (1-2 h) y tienen una semivida de 10-14 h, lo que permite su administración una vez al día. En dosis entre 15 y 150 mg, el edoxabán tiene una farmacocinética lineal y, a diferencia de otros anti-Xa, se une poco a las proteínas plasmáticas y no se biotransforma a través del CYP3A4, lo que podría traducirse en menos riesgo de interacciones farmacológicas.
Conflicto de interesesNinguno.