
La muerte súbita cardiaca (MSC) de origen no isquémico está causada predominantemente por miocardiopatías y canalopatías. La batería de test diagnósticos es amplia e incluye pruebas complejas. El objetivo de nuestro estudio es analizar la rentabilidad diagnóstica del estudio etiológico sistematizado de la MSC.
MétodosSe estudió a 56 familias con al menos 1 caso índice con MSC (reanimada o no). En los supervivientes se exploró con electrocardiograma, imagen cardiaca avanzada, ergometría, estudio familiar, estudio genético y, puntualmente, test farmacológicos. En los fallecidos se examinó la necropsia, así como la autopsia molecular con next generation sequencing (NGS), junto con estudio clínico familiar.
ResultadosEl diagnóstico se alcanzó en el 80,4% de los casos, sin diferencias entre supervivientes y fallecidos (p=0,53). Entre los supervivientes, el diagnóstico de canalopatía fue más frecuente que entre los fallecidos (el 66,6 frente al 40%; p=0,03). De los 30 sujetos fallecidos, en 7 la autopsia aportó un hallazgo concluyente. El diagnóstico de miocardiopatía tendía a asociarse con mayor tasa de eventos en la familia. El test genético con NGS se realizó en 42 de los casos; se obtuvo resultado positivo en 28 (66,6%), sin diferencias entre supervivientes y fallecidos (p=0,21).
ConclusionesLa probabilidad de alcanzar el diagnóstico en la MSC tras un protocolo exhaustivo es alta, con mayor prevalencia de canalopatías en los supervivientes y un aparente peor pronóstico en las miocardiopatías. El test genético mediante NGS muestra utilidad en casos de MSC e incrementa la rentabilidad respecto al estudio con Sanger.
Palabras clave
La muerte súbita cardiaca (MSC) se debe, en la mayoría de los casos, a una arritmia ventricular fatal como la fibrilación ventricular que conduce a la muerte en pocos minutos. Su etiología más frecuente es la cardiopatía isquémica, pero en sujetos menores de 35 años las causas más comunes son las miocardiopatías y las canalopatías de origen genético1–3. Esta etiología genética es cada vez más compleja, con gran heterogeneidad y número de genes involucrados4,5. Diversos trabajos han estudiado la rentabilidad de protocolos diagnósticos para el estudio de la etiología de la MSC, entre ellas técnicas convencionales, como el electrocardiograma (ECG) o el ecocardiograma, y otras más complejas, como la resonancia magnética cardiaca, los test farmacológicos y el estudio genético6–12.
Pocos estudios han analizado la rentabilidad diagnóstica de los diferentes test en función de la supervivencia a la parada cardiaca. Además, hay escasa información de la utilidad del test genético mediante next generation sequencing (NGS) en casos seleccionados de MSC (autopsia molecular), y la penetrancia familiar de dicha etiología genética10–18. Nuestro objetivo es evaluar la rentabilidad diagnóstica de un protocolo amplio, con el uso escalonado de test convencionales, y otros complejos como el estudio genético mediante NGS en casos seleccionados, en una muestra de sujetos con MSC, supervivientes o no supervivientes al evento de parada cardiaca, así como valorar la penetrancia de dichas enfermedades en la familia.
MÉTODOSSe analizaron de manera retrospectiva casos de MSC no isquémica y se estudiaron en una consulta especializada. El criterio de inclusión fue haber padecido una parada cardiaca que precisase desfibrilación externa para restaurar ritmo sinusal en los casos supervivientes. En los sujetos fallecidos, que la muerte ocurriese en la primera hora tras el inicio de los síntomas o antes de 24 h desde la última vez que se constató que el sujeto estaba con vida, en ausencia de causa extracardiaca. Entre los criterios de exclusión se incluyeron el origen isquémico del evento, la causa secundaria evidente de MSC y la negativa a participar en el registro del probando/familia. Un caso se consideró como de etiología isquémica si se demostraron datos de oclusión coronaria aguda en el ECG o coronariografía o ante la presencia de escara de necrosis en las pruebas de imagen en el sujeto superviviente, o por el hallazgo de datos de infarto de miocardio previo con escara de necrosis, el hallazgo de trombo intracoronario oclusivo o la presencia de placas de ateroma en 1 o más arterias coronarias con oclusión de la luz>75% en el sujeto fallecido. El diseño del estudio fue longitudinal y lo aprobó el comité ético local.
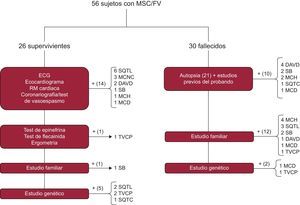
El protocolo diagnóstico aplicado se resume en la figura 116. En el caso del sujeto superviviente, la batería de pruebas se centraba en el caso índice. Aparte de una anamnesis para identificación de factores desencadenantes, toma de fármacos, fiebre o eventos previos, en un primer escalón se realizaban estudios convencionales como ECG basal y de esfuerzo, ecocardiograma transtorácico, coronariografía con y sin test de vasoespasmo y técnicas de imagen cardiaca avanzada. Si no se hallaba el diagnóstico, se aplicaba el protocolo secuencial descrito por nuestro grupo en el estudio FIVI-Gen9, para desenmascarar una canalopatía oculta. Brevemente, se practicaban inicialmente test farmacológicos de epinefrina y flecainida, seguidos de estudio a familiares de primer grado con ECG o ecocardiograma, y si todo resultaba negativo, se estudiaba genéticamente al caso índice.

Algoritmo diagnóstico ante un caso de MSC, tanto en supervivientes como en fallecidos. DAVD: displasia arritmogénica de ventrículo derecho; ECG: electrocardiograma; FV: fibrilación ventricular; MCD: miocardiopatía dilatada; MCH: miocardiopatía hipertrófica; MCNC: miocardiopatía no compactada; MSC: muerte súbita cardiaca; RM: resonancia magnética; SB: síndrome de Brugada; SQTC: síndrome de QT corto; SQTL: síndrome de QT largo; TVPC: taquicardia ventricular polimórfica catecolaminérgica.
Para el caso del sujeto fallecido, se tomaron datos clínicos sobre las circunstancias de la muerte, ritmo de la parada cardiaca, eventos y estudios previos. La necropsia se realizó por el Instituto de Medicina Legal de Granada en la mayoría de los casos, siguiendo las guías y recomendaciones actuales19. En resumen, se realizaba un examen macroscópico del corazón, y se registraban peso, tamaño y grosor de cavidades, origen, trayecto y luz de arterias coronarias, presencia de trombosis coronaria o datos de isquemia miocárdica aguda, anatomía valvular y descripción macroscópica y microscópica del miocardio. Se realizó estudio bioquímico, toxicológico e histológico en todos los casos disponibles, y en los casos sin diagnóstico cierto o con diagnóstico familiar cierto, pero a juicio del investigador se realizó autopsia molecular (figura 1 y anexo 1 del material suplementario) o se secuenció genéticamente a un familiar directo con evidencia de la enfermedad si no había ADN disponible del fallecido.
El estudio familiar se realizó en cascada en todos los casos en que se llegó a un diagnóstico clínico o genético.
Estudio genéticoEl estudio genético se realizó en la mayoría de familias, hubiese o no diagnóstico clínico. Se excluyeron algunos casos con diagnóstico de una miocardiopatía o canalopatía en los que la genética aporta poca rentabilidad clínica y no hay evidente agregabilidad familiar, como el síndrome de Brugada, la miocardiopatía no compactada y algunos casos de miocardiopatía dilatada. El ADN se obtuvo de muestras de sangre periférica almacenada con EDTA. Se evaluaron una serie de genes (hasta 242) previamente asociados con miocardiopatías y canalopatías. La secuenciación de estos se realizó mediante método Sanger en los estudios realizados antes de 2013, y desde entonces, mediante la plataforma NGS Illumina 1500 Hiseq al estar disponible esta tecnología (véase los detalles técnicos en el anexo 2 del material suplementario). Casi todos los casos que fueron negativos con tecnología Sanger se volvieron a secuenciar con NGS posteriormente. Se obtuvo la secuencia de todas las regiones exónicas codificantes e intrónicas flanqueantes. La patogenicidad de las variantes detectadas se catalogó según la descripción previa de la variante, la predicción in silico de patogenicidad por las herramientas bioinformáticas (Polyphen20, SIFT21, MutationTaster22), el grado de conservación del residuo a través de las especies y su presencia en bases de datos públicas de la población general como dbSNP y la NHLBI GO Exome Sequencing Project database. Finalmente, para considerar una mutación como probablemente patogénica, se requirió una coherencia y cosegregación familiar con el fenotipo.
Análisis estadísticoSe creó una base de datos específica para el estudio. Se empleó para el análisis de resultados el programa estadístico SPSS (versión 20.0) (STATA Corp.; Chicago, Estados Unidos). Para la comparación de variables cuantitativas se empleó la prueba de la U de Mann-Whitney y para el estudio de proporciones, el test exacto de Fisher. Se consideró significativo un valor de p<0,05.
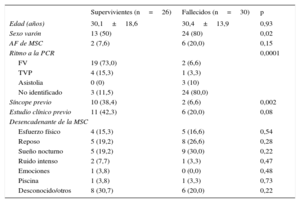
RESULTADOSLa muestra final consistió en 56 casos índice o familias (edad media de los casos, 30,2±16,1 años; el 66,1% varones). El 53,6% eran probandos fallecidos (30 casos) frente al resto, que eran supervivientes (26 casos índice). Para el caso concreto de los fallecidos, los casos enviados fueron aquellos en los que se sospechó cardiopatía familiar entre una media de 145 episodios anuales de MSC durante el periodo de estudio, de los cuales, revisados retrospectivamente, hasta un 15% aproximadamente podría deberse a una cardiopatía familiar. Hubo una mayor prevalencia de varones en el grupo de fallecidos, sin diferencias en la edad u otras variables sociodemográficas (tabla 1). Se realizó un estudio cardiológico previo al 30,4% de los casos, fundamentalmente por haber sufrido un síncope (el 21,4%).
Características sociodemográficas de los casos de muerte súbita cardiaca en supervivientes y fallecidos
| Supervivientes (n=26) | Fallecidos (n=30) | p | |
|---|---|---|---|
| Edad (años) | 30,1±18,6 | 30,4±13,9 | 0,93 |
| Sexo varón | 13 (50) | 24 (80) | 0,02 |
| AF de MSC | 2 (7,6) | 6 (20,0) | 0,15 |
| Ritmo a la PCR | 0,0001 | ||
| FV | 19 (73,0) | 2 (6,6) | |
| TVP | 4 (15,3) | 1 (3,3) | |
| Asistolia | 0 (0) | 3 (10) | |
| No identificado | 3 (11,5) | 24 (80,0) | |
| Síncope previo | 10 (38,4) | 2 (6,6) | 0,002 |
| Estudio clínico previo | 11 (42,3) | 6 (20,0) | 0,08 |
| Desencadenante de la MSC | |||
| Esfuerzo físico | 4 (15,3) | 5 (16,6) | 0,54 |
| Reposo | 5 (19,2) | 8 (26,6) | 0,28 |
| Sueño nocturno | 5 (19,2) | 9 (30,0) | 0,22 |
| Ruido intenso | 2 (7,7) | 1 (3,3) | 0,47 |
| Emociones | 1 (3,8) | 0 (0,0) | 0,48 |
| Piscina | 1 (3,8) | 1 (3,3) | 0,73 |
| Desconocido/otros | 8 (30,7) | 6 (20,0) | 0,22 |
AF: antecedente familiar; FV: fibrilación ventricular; MSC: muerte súbita cardiaca; PCR: parada cardiorrespiratoria; TVP: taquicardia ventricular polimórfica.
Los valores expresan n (%) o media±desviación estándar.
El diagnóstico final se obtuvo en un 80,4% de las familias, pero no se encontraron diferencias estadísticamente significativas en función del grupo de estudio (supervivientes frente a fallecidos, el 80,7 y el 80%; p=0,53). La figura 1 muestra cómo se alcanzó el diagnóstico definitivo en ambos grupos. El ECG estaba disponible en el 100% de los casos índice supervivientes, frente al 23,3% de los fallecidos. Hubo diferencias significativas en la forma de diagnosticar a cada familia en función del grupo de estudio, ya que en el grupo de fallecidos la contribución del estudio familiar apoyado en los datos de la autopsia fue la clave de la mayoría de los casos, mientras que en los supervivientes el diagnóstico se alcanzó sobre todo a base de pruebas clínicas realizadas al probando. No obstante, el estudio familiar aportó datos fundamentales para lograr el diagnóstico en un caso con FV idiopática en el probando y test de provocación farmacológicos positivos en familiares. La autopsia se realizó en el 70% de los casos, y se consideró concluyente (bien por la identificación de una miocardiopatía causal [11 casos], bien por ser «blanca» [5 casos]) en el 76% de los casos. En 5 casos la autopsia mostró datos dudosos, borderline o incompletos. Realizó la autopsia el Instituto de Medicina Legal de Granada en 15 casos, mientras que en 6 la llevó a cabo el Instituto de Medicina Legal de la ciudad en la que ocurrió el fallecimiento y nosotros realizamos el estudio familiar.
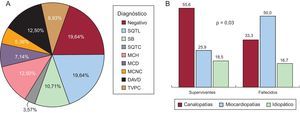
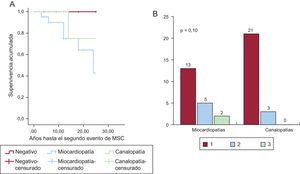
La figura 2 muestra una representación esquemática de las enfermedades halladas, y su prevalencia en función del grupo de estudio. La prevalencia de canalopatías fue superior de manera estadísticamente significativa entre los supervivientes, fundamentalmente a expensas del síndrome de QT largo (8 casos). Una proporción similar en ambos grupos quedó finalmente sin diagnóstico definitivo; la frecuencia de diagnóstico de miocardiopatía fue significativamente superior en los fallecidos, fundamentalmente a expensas de miocardiopatía hipertrófica y arritmogénica. Además, la presencia de un diagnóstico de miocardiopatía se relacionó, aunque de manera no estadísticamente significativa, con la probabilidad de que se presentara un segundo evento arrítmico en la familia, estudiado desde un punto de vista retrospectivo. La figura 3 muestra una curva de supervivencia para la presencia de un segundo evento arrítmico en la familia, bien una MSC, bien una arritmia ventricular o un síncope. La probabilidad de que se presente más de un evento arrítmico maligno fue del 36,8% en las familias con miocardiopatía y del 12,1% en las familias con canalopatías (p=0,10). Los sujetos diagnosticados de canalopatía eran significativamente más jóvenes que aquellos con miocardiopatía (26±16,7 frente al 36,1±16 años; p=0,04).

A: diagnósticos obtenidos en general. B: comparación entre ambos grupos. DAVD: displasia arritmogénica de ventrículo derecho; MCD: miocardiopatía dilatada; MCH: miocardiopatía hipertrófica; MCNC: miocardiopatía no compactada; SB: síndrome de Brugada; SQTC: síndrome de QT corto; SQTL: síndrome de QT largo; TVPC: taquicardia ventricular polimórfica catecolaminérgica.

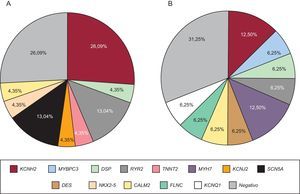
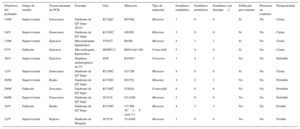
Se realizó test genético al 71,4% de los casos, bien al probando, bien a familiares directos con fenotipo positivo, en caso de no disponerse de ADN del sujeto fallecido. El test genético se realizó dirigido por fenotipo, bien por NGS con paneles concretos seleccionados de genes candidatos (28 casos, 9 de los cuales se realizaron previamente estudio con Sanger) o exclusivamente mediante la técnica de Sanger (12 casos). El test genético fue positivo con el hallazgo de una probable mutación causal en el 67,5% de los casos, pero no se apreciaron diferencias estadísticamente significativas en función del grupo de estudio ni en relación con el método de secuenciación empleado. Los genes más frecuentemente mutados en ambos grupos fueron KCNH2, RyR2 y DSP (figura 4). Se estudió en cascada a un total de 298 familiares, una media de 5,2 por familia, que permitió la identificación de 113 sujetos afectados. La figura 5 muestra un ejemplo de estudio familiar en cascada. La tabla 2 y la tabla 3 detallan los casos de familias con resultado genético positivo obtenido mediante NGS y Sanger respectivamente. En 9 de los casos realizados mediante NGS, se había realizado previamente un estudio con Sanger que resultó negativo, y permitió el diagnóstico de 5 de estos casos con el hallazgo de una mutación causal. En conjunto, el test genético fue la clave para el diagnóstico del 12,2% de las familias, y mostró tendencia a mayor rentabilidad en los casos de canalopatías que en las miocardiopatías (el 95 frente al 69% de positividad; p=0,08).
 y fallecidos (B).")
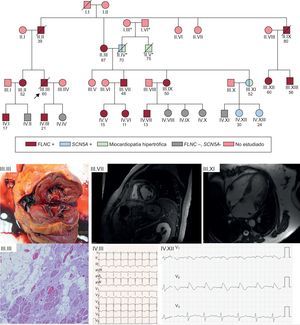
, causante de síndrome de Brugada y patogénica. En las figuras clínicas de la parte inferior de la imagen se aprecia: III.III: imagen macroscópica de corte de ventrículo izquierdo de pieza tras el fallecimiento, con dilatación del ventrículo y surco mesocárdico de fibrosis. En la imagen microscópica, fibrosis abundante y reemplazo de miocardiocitos con tejido adiposo. III.VII: varón de 48 años portador de mutación en FLNC con fibrosis extensa mesocárdica de ventrículo izquierdo, hallazgos similares a los observados en la autopsia de III.III. IV.III: ECG de paciente de 21 años afectado por mutación en FLNC y miocardiopatía de ventrículo izquierdo. III.XI: miocardiopatía hipertrófica en mujer de 52 años no portadora de la mutación en FLNC, pero sí mutación Glu1225Lys en SCN5A. IV.XII: patrón de Brugada tipo 1 tras flecainida en hija asintomática de la paciente previa. ECG: electrocardiograma; MSC: muerte súbita cardiaca; NGS: next generation sequencing. *: pertenecientes a una familia diferente a la que motivó el estudio; /: sujeto fallecido; ○: mujeres; □: varones; : probando.")
Ejemplo de familia con mutación en FLNC p.Leu194fs y alta tasa de eventos de MSC. El estudio en cascada permitió la identificación de 14 miembros afectados por la misma mutación, con cosegregación muy elevada para el fenotipo de miocardiopatía arritmogénica. El estudio en cascada permitió el hallazgo casual de miocardiopatía hipertrófica en una familia no afectada por la mutación en FLNC, pero que en el estudio con NGS portaba otra mutación en SCN5A heredada por el otro ramo de la familia (su padre), causante de síndrome de Brugada y patogénica. En las figuras clínicas de la parte inferior de la imagen se aprecia: III.III: imagen macroscópica de corte de ventrículo izquierdo de pieza tras el fallecimiento, con dilatación del ventrículo y surco mesocárdico de fibrosis. En la imagen microscópica, fibrosis abundante y reemplazo de miocardiocitos con tejido adiposo. III.VII: varón de 48 años portador de mutación en FLNC con fibrosis extensa mesocárdica de ventrículo izquierdo, hallazgos similares a los observados en la autopsia de III.III. IV.III: ECG de paciente de 21 años afectado por mutación en FLNC y miocardiopatía de ventrículo izquierdo. III.XI: miocardiopatía hipertrófica en mujer de 52 años no portadora de la mutación en FLNC, pero sí mutación Glu1225Lys en SCN5A. IV.XII: patrón de Brugada tipo 1 tras flecainida en hija asintomática de la paciente previa. ECG: electrocardiograma; MSC: muerte súbita cardiaca; NGS: next generation sequencing. *: pertenecientes a una familia diferente a la que motivó el estudio; /: sujeto fallecido; ○: mujeres; □: varones;
: probando.
Familias con diagnóstico genético obtenido mediante next generation sequencing
| Edad/sexo del probando | Grupo de estudio | Desencadenante de PCR | Fenotipo | Gen | Mutación | Tipo de mutación | Familiares estudiados | Familiares portadores | Familiares con fenotipo+ | Publicada previamente | Presencia en controles | Patogenicidad |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 15/M | Superviviente | Sueño | Miocardiopatía no compactada+cardiopatía congénita | NKX2-5 | E167K | Missense | 7 | 2 | 2 | No | No | Probable |
| 17/V | Superviviente | Piscina | FVI-SQTC | KCNH2 | R1035W | Missense | 3 | 1 | 0 | Sí | <1% | Posible |
| 45/V | Fallecido | Ejercicio | Miocardiopatía hipertrófica | MYH7 | R694H | Missense | 1 | 1 | 1 | Sí | No | Cierta |
| 15/V | Superviviente | Reposo | Síndrome de QT largo | KCNQ1+KCNH2 | S644M.fs + V115M | Frameshift | 3 | 1 | 1 | No | No | Cierta |
| 50/V | Fallecido | Ejercicio | Miocardiopatía arritmogénica de VI | DSP | R2284X | Missense | 8 | 4 | 4 | Sí | No | Cierta |
| 31/V | Fallecido | Reposo | Miocardiopatía arritmogénica de VI | DES | E401D | Missense | 66 | 23 | 22 | No | No | Probable |
| 23/M | Superviviente | Emociones | Taquicardia ventricular catecolaminérgica | RYR2 | A2387T | Missense | 4 | 1 | 1 | SÍ | No | Probable |
| 16/V | Superviviente | Emociones | Taquicardia ventricular catecolaminérgica | RYR2 | K337N | Missense | 3 | 2 | 0 | No | No | Probable |
| 15/V | Superviviente | Ejercicio físico | Taquicardia ventricular catecolaminérgica | RYR2 | L4915W | Missense | 3 | 0 | 0 | No | No | Probable |
| 40/M | Superviviente | Reposo | Síndrome de Andersen Tawil (SQTL tipo 7) | KCNJ2 | C122Y | Missense | 2 | 1 | 1 | No | No | Probable |
| 53/M | Superviviente | Reposo | FVI-síndrome de Brugada | SCN5A | D1816.fs | Frameshift | 14 | 7 | 3 | No | No | Probable |
| 7/V | Fallecido | Ejercicio | Muerte súbita inexplicada | CALM2 | N98S | Missense | 3 | 0 | 0 | No | No | Probable |
| 16/V | Fallecido | Piscina | Taquicardia ventricular catecolaminérgica | RyR2 | L488I | Missense | 5 | 4 | 4 | No | No | Probable |
| 4/M | Superviviente | Ruido | FVI-síndrome de QT largo | CALM2 | N98S | Missense | 3 | 0 | 0 | No | No | Probable |
| 60/V | Fallecido | Sueño | Miocardiopatía arritmogénica de VI | FLNC | L194.fs | Frameshift | 23 | 14 | 12 | No | No | Probable |
| 43/V | Fallecido | Sueño | Síndrome de QT largo | KCNQ1 | R366Q | Missense | 16 | 10 | 10 | Sí | No | Probable |
FVI: fibrilación ventricular idiopática; M: mujer; PCR: parada cardiorrespiratoria; SQTC: síndrome de QT corto; V: varón; VI: ventrículo izquierdo.
Familias con diagnóstico genético obtenido mediante Sanger
| Edad/sexo del probando | Grupo de estudio | Desencadenante de PCR | Fenotipo | Gen | Mutación | Tipo de mutación | Familiares estudiados | Familiares portadores | Familiares con fenotipo+ | Publicada previamente | Presencia en controles | Patogenicidad |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 15/M | Superviviente | Emociones | Síndrome de QT largo (JLN) | KCNQ1 | R539Q | Missense | 3 | 0 | 0 | Sí | No | Cierta |
| 18/V | Superviviente | Emociones | Síndrome de QT largo | KCNH2 | G628S | Missense | 3 | 0 | 0 | Sí | No | Cierta |
| 13/M | Superviviente | Ejercicio | Miocardiopatía hipertrófica | TNNT2 | R94H | Missense | 4 | 1 | 1 | Sí | No | Cierta |
| 57/V | Fallecido | Ejercicio | Miocardiopatía hipertrófica | MYBPC3 | R891Afs*160 | Frameshift | 5 | 4 | 4 | Sí | No | Cierta |
| 36/V | Superviviente | Ejercicio | Displasia arritmogénica de VI | DSP | R1045* | Nonsense | 3 | 2 | 1 | No | No | Probable |
| 11/V | Superviviente | Emociones | Síndrome de QT largo | KCNH2 | G572R | Missense | 4 | 0 | 0 | Sí | No | Cierta |
| 49/M | Superviviente | Ruido | Síndrome de QT largo | KCNH2 | E637G | Missense | 4 | 2 | 2 | No | No | Probable |
| 29/M | Fallecido | Descanso | Síndrome de QT largo | KCNH2 | G262fs | Frameshift | 8 | 6 | 6 | No | No | Probable |
| 60/M | Superviviente | Emociones | Síndrome de QT largo | SCN5A | G1329S | Missense | 2 | 0 | 0 | No | No | Probable |
| 25/V | Fallecido | Ruido | Síndrome de QT largo | KCNH2 | 5’UTR-4C>T (isof. C) | Missense | 4 | 3 | 3 | No | No | Posible |
| 22/V | Superviviente | Reposo | Síndrome de Brugada | SCN5A | Y1449S | Missense | 3 | 0 | 0 | No | No | Posible |
JLN: síndrome de Jervell y Lange-Nielsen; M: mujer; V: varón; VI: ventrículo izquierdo.
Nuestro trabajo muestra una elevada rentabilidad diagnóstica aplicando un protocolo sistemático para casos de MSC. No hubo diferencias en la tasa final de diagnósticos obtenidos en función de la supervivencia a la parada cardiaca ni en sus desencadenantes, pero sí en la forma de alcanzar el diagnóstico, con una elevada rentabilidad del estudio necrópsico y familiar en las familias de los fallecidos, mientras que entre los supervivientes la forma de alcanzar el diagnóstico fue más heterogénea y con técnicas convencionales aplicadas sistemáticamente al probando. El test genético mostró una alta tasa de resultados positivos; aunque no significativamente, la tecnología NGS se mostró superior a Sanger.
Hay pocos estudios que analicen las características diferenciales de los casos de MSC en función de la supervivencia a lsa parada cardiaca. Un grupo australiano11 encontró diferencias en la rentabilidad diagnóstica entre fallecidos y supervivientes, con un porcentaje de diagnósticos 3,5 veces superior entre estos. En ese trabajo, a diferencia del nuestro, se excluyeron los casos de cardiopatía estructural. Para el diagnóstico de canalopatías es fundamental disponer del probando superviviente para la realización de ECG basal y con epinefrina, flecainida o esfuerzo. Nuestros datos concuerdan con esta observación, pues la prevalencia de canalopatías fue superior en el grupo de supervivientes. En nuestro trabajo destaca la elevada rentabilidad diagnóstica de la autopsia junto con estudio familiar para el diagnóstico de miocardiopatías, que permitió el diagnóstico en la mayoría de los casos. Este dato difiere de series previas, y puede ser atribuible a varios motivos. Por un lado, estudiamos a un amplio número de familiares por caso índice (5,2), superior al de series previas como la australiana (con 3,8), la serie de Behr et al.17 (con 3,2) o el registro CASPER10 (con 1,3). Además, el carácter retrospectivo de nuestra serie podría suponer un sesgo de selección, pues se estudiaron preferentemente los casos con hallazgos patológicos evidentes en la autopsia.
La mayor prevalencia de miocardiopatías en el grupo de fallecidos podría indicar un peor pronóstico de la parada cardiaca en los pacientes con miocardiopatías respecto a los trastornos eléctricos primarios. Por un lado, el mecanismo desencadenante de la parada en las canalopatías pudiera ser distinto que en las miocardiopatías y, por otro, es posible que el miocardio normal de las canalopatías sea menos vulnerable a la isquemia y facilite la respuesta a las maniobras de reanimación y desfibrilación. Respalda este hecho que más pacientes del grupo de supervivientes padecieran síncope, mientras que en los sujetos fallecidos con alta prevalencia el primer evento fue la MSC. Además, la tasa de eventos en familiares fue mayor entre los pacientes con miocardiopatías, con mayor prevalencia de MSC y tendencia a menor tiempo entre dichos eventos. Estos datos ya se describieron en el seguimiento de los pacientes del registro CASPER10, donde se apreció una mayor frecuencia de terapias apropiadas en el grupo de pacientes con miocardiopatías, y se propone un comportamiento más agresivo que para aquellos con trastornos eléctricos primarios23.
Las características diferenciales entre ambos grupos también mostraron un dato concordante con la literatura previa24 y es el mayor porcentaje de varones en el total de la serie, y en particular dentro del grupo de fallecidos. No hubo diferencias entre los factores precipitantes de la parada cardiaca, con escasa incidencia del ejercicio intenso o el deporte de competición. Este dato es relevante, ya que actualmente se recomienda abandonar el ejercicio físico de competición a los sujetos diagnosticados de miocardiopatía, con algo de mayor permisividad en las canalopatías, y su papel podría no ser tan crucial25. Además, el hecho de que los factores precipitantes de la parada cardiaca sean similares resta importancia a la posibilidad de que pertenecer a un grupo u otro dependa de las circunstancias en que se produzca la muerte.
Un factor que contribuyó de manera significativa a aumentar la rentabilidad diagnóstica en ambos grupos fue el estudio genético. Si bien el número total de casos en los cuales fue útil para alcanzar el diagnóstico definitivo fue discreto, su uso y su interpretación adecuados permitieron la identificación de gran número de familiares afectados. Estudios previos analizaron la rentabilidad del test genético dirigido por fenotipo o en casos sin fenotipo aparente8–12. En el caso de la autopsia molecular, clásicamente se ha recomendado la secuenciación de pocos genes, como KCNQ1, KCNH2, SCN5A y RyR214,26, con ampliación en casos concretos a algunas miocardiopatías para facilitar el estudio familiar en cascada. En el caso de sujetos supervivientes a parada cardiaca en ausencia de fenotipo, este estudio está incluso desaconsejado por las guías de práctica clínica27. Un estudio previo de nuestro grupo indicó que el uso de la nueva tecnología NGS aplicada a sujetos supervivientes a parada cardiaca en ausencia de fenotipo podría aumentar la rentabilidad diagnóstica9. En el presente trabajo se aporta evidencia, además, de que dicho test genético realizado mediante NGS en sujetos tanto supervivientes como fallecidos (autopsia molecular) permite identificar casos de mutaciones patogénicas en genes poco habituales, como CALM, FLNC, DES, etc., y que permiten el diagnóstico familiar en cascada. La tecnología NGS supone una oportunidad en el ámbito de la MSC por su potencial diagnóstico para las miocardiopatías y canalopatías28, y debe ser la técnica de secuenciación genética empleada en el abordaje de la MSC29. Dada su capacidad para simplificar el análisis de numerosos genes y aumentar la sensibilidad respecto a Sanger, el reto es la correcta interpretación de los hallazgos para no perder especificidad, para lo cual es crucial un estudio de cosegregación amplio como el llevado a cabo en nuestra serie. Un trabajo muy reciente, prospectivo y con gran muestra de pacientes cifra en el 27% la utilidad real de la autopsia molecular mediante NGS en casos de MSC no explicada12.
Limitaciones del estudioAunque se aplicó un protocolo de estudio sistemático a los casos enviados a consulta especializada, el diseño retrospectivo del estudio sugiere que algunos casos, sobre todo los fallecidos, podrían haberse perdido por no haber un protocolo establecido de derivación y ciertos casos podrían estar seleccionados en función de los hallazgos sospechosos de cardiopatía familiar más evidentes en la autopsia. Esto explica la diferencia entre los casos fallecidos incluidos y los que serían esperables. Además, esta es la razón de no disponer del 100% de los estudios necrópsicos; no todos los estudios necrópsicos se realizaron por el Instituto de Medicina Legal de Granada, con lo que se podría incurrir en cierta variabilidad interobservadores. Este carácter retrospectivo de la recogida de los estudios necrópsicos motivó que 5 casos no tuviesen datos concluyentes y que no se pudiese ampliar el estudio tras el fallecimiento. Este hecho se minimiza con la interpretación rigurosa y unicéntrica de los informes necrópsicos y el estudio centralizado y homogéneo realizado de todas las familias en nuestra unidad. Otra limitación es el tamaño muestral, que podría resultar insuficiente para obtener conclusiones más sólidas.
CONCLUSIONESLa probabilidad de alcanzar el diagnóstico final en la MSC tras un protocolo exhaustivo es alta, con mayor prevalencia de canalopatías en los supervivientes y un aparente peor pronóstico en las familias con miocardiopatías y en varones. El test genético mediante NGS muestra utilidad en casos de MSC con y sin fenotipo aparente, lo que incrementa la rentabilidad respecto al estudio de genes puntuales con Sanger.
CONFLICTO DE INTERESESNinguno.
- –
La MSC no isquémica se debe a enfermedades hereditarias que hacen posible la existencia de casos ocultos en la familia.
- –
El sexo masculino se relaciona con mayor prevalencia de MSC. No está demostrado un peor pronóstico familiar en función del diagnóstico subyacente.
- –
Los test farmacológicos, el estudio familiar y la autopsia molecular tienen un papel en el estudio de la MSC no aclarada.
- –
La utilidad del test genético en casos con fenotipo evidente está demostrada con técnicas convencionales, pero no hay suficiente evidencia respecto a la tecnología NGS en el estudio de la MSC.
- –
Con un estudio sistematizado de probando y familia se alcanza el diagnóstico definitivo en un alta proporción de casos de MSC.
- –
La incidencia de casos entre los familiares es alta. Un estudio familiar riguroso puede ser la clave para alcanzar el diagnóstico tanto en supervivientes como fallecidos por MSC.
- –
En las familias con un caso de MSC, el diagnóstico de miocardiopatías parece conllevar mayor tasa de eventos y peor pronóstico.
- –
El test genético con NGS parece ser superior a la tecnología Sanger por su capacidad para detectar mutaciones en genes poco habituales.
En primer lugar, agradecemos a las familias participantes por su generosidad en un momento de la vida tan trágico. También queremos agradecer a la Dra. C. Moro, del Instituto de Medicina Legal de Sevilla, su valiosa contribución para el estudio histológico de las muestras.