Palabras clave
INTRODUCCIÓN
La diabetes mellitus (DM) es una enfermedad crónica cuya morbilidad y mortalidad a largo plazo deriva de las consecuencias del desarrollo de enfermedad vascular aterosclerótica. La DM tipo 2 es la forma más frecuente y su prevalencia está aumentando de manera paralela al incremento de la edad poblacional y a la incidencia de la obesidad e inactividad física en las sociedades de países desarrollados1. La cardiopatía isquémica es la principal causa de muerte en pacientes con DM tipo 22.Existen evidencias de que los pacientes diabéticos que no han sufrido un síndrome coronario agudo tienen el mismo riesgo de padecerlo que aquellos no diabéticos que lo han sufrido previamente3,4. Esto sugiere que, subyacente a la DM, puede existir una extensa aterosclerosis coronaria sin una obvia manifestación clínica.Además, el riesgo de muerte por un acontecimiento cardiovascular en los pacientes diabéticos es dos-cuatro veces mayor que en la población no diabética5,6. Este exceso de mortalidad es más elevado en mujeres (cuatro-cinco veces) que en varones (dos-tres veces)3,7. En los últimos años se ha constatado una disminución en la mortalidad de causa coronaria debido probablemente a los avances en el tratamiento médico, como el uso de agentes fibrinolíticos8, antiagregantes9, estatinas10, bloqueadores beta11, insulinoterapia postinfarto de miocardio12 y al mejor control de la hiperglucemia13. Sin embargo, esta disminución ha tenido menos impacto en pacientes diabéticos14. Por estos motivos, la American Heart Association considera la DM no como un simple factor independiente de riesgo, sino como una «verdadera enfermedad cardiovascular»1.
La enfermedad coronaria en los pacientes diabéticos suele ser de naturaleza difusa y ocasiona unas arterias coronarias no óptimas para la revascularización. Otra de las características es que por lo general progresa con rapidez, por lo que tanto las angioplastias con o sin stent como los injertos aortocoronarios suelen tener una vida media más corta en pacientes diabéticos que en no diabéticos15-17.Las causas de esta mayor severidad y rápida progresión en enfermos diabéticos no se han esclarecido por completo4.
En este artículo se revisan los posibles mecanismos de aterogénesis y enfermedad coronaria en la DM y se analiza el potencial beneficio de nuevos fármacos con acción antiinflamatoria y sensibilizante a la insulina.
RESISTENCIA A LA INSULINA, SÍNDROME METABÓLICO Y ATEROSCLEROSIS
El mecanismo fisiopatogénico de la DM tipo 2 es en la actualidad poco conocido. Se postula la hipótesis de la combinación de un déficit de secreción de insulina por las células beta pancreáticas con una resistencia periférica a la acción de la insulina. La secreción de insulina disminuye con la edad y puede verse acelerada por factores genéticos18. La resistencia a la insulina es una anormalidad celular compleja que implica a varios órganos, especialmente al tejido adiposo, al hígado y al músculo esquelético, y que predispone a varios defectos metabólicos19,20.Hoy día se cree que la resistencia a la insulina se desarrolla como consecuencia de la interacción de factores ambientales (como los mencionados obesidad e inactividad física) y factores genéticos1. De manera característica, la resistencia a la insulina precede en años al comienzo de la DM tipo 221,22 y suele acompañarse de otros factores de riesgo cardiovascular como la dislipemia, la obesidad, la hipertensión y un estado protrombótico. En 1988, Reaven describió el «síndrome X» como el conjunto de factores de riesgo metabólico que tienden a ocurrir en el mismo individuo y que pueden desempeñar un papel fundamental en el desarrollo de la enfermedad coronaria23. El elemento común y subyacente del síndrome es la resistencia a la insulina, mientras que el desarrollo de la DM tipo 2 y la enfermedad coronaria se postulan como alteraciones secundarias23-26. El síndrome metabólico es una entidad epidemiológica o una tendencia, y no un síndrome clínico que pueda ser diagnosticado y tratado27. La importancia del síndrome X metabólico radica en que su descripción ha abierto nuevos caminos para la investigación de nuevos factores de riesgo cardiovascular. Actualmente, como componentes mayores del síndrome metabólico se consideran la dislipemia típica de esta entidad, el incremento de la presión arterial, la hiperglucemia y un estado protrombótico20.
Dislipemia
Los tres componentes de la «dislipemia aterogénica» de la resistencia a la insulina son: incremento de las concentraciones de triglicéridos, descenso de las lipoproteínas de alta densidad (HDL) y lipoproteínas de baja densidad (LDL) densas y de tamaño reducido. De nuevo nos encontramos ante una asociación epidemiológica, aunque es probable que exista un defecto metabólico subyacente y común, ya que es raro encontrarlas separadas en personas insulinorresistentes28,29. Se ha postulado que el trastorno inicial en la resistencia a la insulina ocurre en el adipocito y consiste en una incapacidad para almacenar ácidos grasos secundaria a una alteración genética. Esto genera un flujo aumentado de ácidos grasos hacia el hígado, con el subsiguiente incremento en la formación de lipoproteínas de muy baja densidad (VLDL)30. Esta sobreproducción de VLDL puede explicar el resto de alteraciones lipídicas características de este síndrome31. El intercambio de triglicéridos y ésteres de colesterol entre las VLDL con las HDL y las LDL puede enriquecer de triglicéridos a estas dos últimas lipoproteínas. La consecuente hidrólisis mediada por la lipoproteinlipasa y la lipasa hepática (la trigliceridemia aumenta la actividad de la lipoproteinlipasa)32 genera partículas de LDL pequeñas y densas, junto con una menor cantidad de HDL33. Además, esta sobrecarga lipídica en el músculo esquelético disminuye la sensibilidad a la insulina y puede explicar la resistencia a ésta33,34.
El estudio de Quebec ha demostrado que la presencia de LDL pequeñas y densas es un factor independiente de riesgo coronario lo que, añadido a las concentraciones elevadas de apo B, puede constituir el estado metabólico que mejor predice el desarrollo de la enfermedad coronaria35. Se especula que estas LDL de menor tamaño son más susceptibles al estrés oxidativo y poseen una mayor capacidad de filtración en la pared arterial36. Se ha establecido que la existencia de una concentración plasmática de HDL disminui da constituye un factor de riesgo coronario independiente37. Lo mismo se cree de las VLDL remanentes (ß-VLDL), que son ricas en colesterol y, por tanto, potencialmente aterogénicas38.
Hipertensión arterial
La hipertensión arterial es otro factor independiente de riesgo cardiovascular y la relación que mantiene con la resistencia a la insulina es una de las cuestiones más controvertidas de este síndrome39. Los mecanismos implicados son el aumento de la actividad del sistema nervioso simpático que ocurre en personas obesas y en aquellas insulinorresistentes40 y el incremento en la reabsorción renal de sodio y agua estimulado por la insulina41; así mismo, últimamente se especula que los ácidos grasos y una acción alterada de la insulina pueden desencadenar una disfunción endotelial dependiente del endotelio42. Como la resistencia a la insulina se asocia al tipo de fibra y a la densidad capilar muscular43, una disfunción endotelial microvascular en el músculo esquelético puede explicar tanto el desarrollo de la hipertensión arterial como la resistencia a la insulina44. El estudio HOPE dejó este interrogante al indicar que el tratamiento con ramipril, agente que mejora la disfunción endotelial, se asoció a un mayor retraso en el inicio de la DM tipo 245.
Alteraciones de la coagulación
La DM predispone a anomalías en la funcionalidad plaquetaria y en los sistemas de coagulación y fibrinolítico que favorecen el proceso trombótico. Factores como el fibrinógeno, el factor VII y el inhibidor tipo 1 del activador del plasminógeno (PAI-1) están aumentados en la sangre de pacientes diabéticos e individuos con resistencia a la insulina46,47. Otros, como la antitrombina III, como resultado de la glicación no enzimatica, presentan una actividad disminuida48. Como comentaremos más adelante, estas alteraciones son fundamentales en la progresión y severidad de la enfermedad coronaria.
Hiperinsulinemia e hiperglucemia
Aunque hoy día se cree que existe una relación entre la hiperglucemia y el desarrollo de enfermedad coronaria49, los estudios de intervención demostraron que la hiperglucemia es un factor de riesgo más importante en el desarrollo de complicaciones microvasculares. Es probable que la dislipemia tenga un papel más importante en la enfermedad macrovascular50,51. Un reciente metaanálisis de 17 estudios prospectivos y de casos y controles consideró la hiperinsulinemia como un débil pero positivo indicador independiente de riesgo cardiovascular52. El Quebec Cardiovascular Study53 demostró una asociación independiente entre la concentración de insulina y la enfermedad coronaria en varones. En el Multiple Risk Factor Intervention Trial54 la hiperinsulinemia resultó ser un factor de riesgo sólo en varones con el fenotipo 3/2 de la apoproteína E. Por tanto, existe controversia en cuanto a si la hiperinsulinemia actúa como marcador del estado de resistencia a la insulina o como verdadero factor de riesgo cardiovascular. La resistencia a la insulina se correlaciona mejor con la enfermedad coronaria que la hiperinsulinemia. En el Insulin Resistance Atherosclerosis Study55 se demostró una asociación significativa entre el grado de aterosclerosis carotídea y la resistencia a la insulina. Sin embargo, esta correlación no se evidenció en individuos de raza negra.
Aunque los epidemiólogos han intentado determinar si esta serie de factores actúan de manera independiente aumentando el riesgo de enfermedad coronaria, la compleja asociación entre los componentes del síndrome metabólico hace que delimitar la independencia de su potencial aterogénico sea extremadamente difícil.
INFLAMACIÓN, DIABETES Y ATEROSCLEROSIS
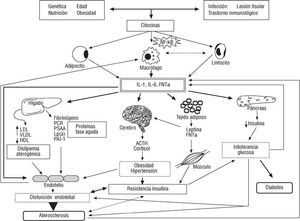
Actualmente existe una gran evidencia de que en el desarrollo y la progresión de la aterosclerosis subyacen mecanismos inmunológicos e inflamatorios56,57. Numerosos estudios demostraron que proteínas de fase aguda como la proteína C reactiva (PCR), el fibrinógeno, la proteína sérica A-amiloide y diversas interleucinas fueron factores predictivos de progresión y severidad de enfermedad coronaria en la población general y en el subgrupo de pacientes diabéticos en particular58-60. Recientes investigaciones han revelado que la inflamación crónica y subclínica parece ser otro componente más del síndrome de resistencia a la insulina. Estudios epidemiológicos han demostrado que la PCR y otros marcadores de inflamación se asocian independientemente con la resistencia a la insulina y con marcadores de disfunción endotelial, etapa inicial del proceso aterogénico61,62. Es importante reseñar el papel que desempeña el tejido adiposo y, por tanto, la obesidad, en el mantenimiento de un estado de inflamación crónico, al secretar una variedad de moléculas biológicamente activas como la interleucina 6 (IL-6), el factor de necrosis tumoral alfa (FNT*), la leptina y la adiponectina, que son determinantes en la regulación del proceso aterogénico y la resistencia a la insulina62-65. Esto nos dirige hacia la hipótesis de que una respuesta inmunológica e inflamatoria sistémica puede subyacer al desarrollo del síndrome metabólico, la DM y la aterosclerosis66 (fig. 1).

Fig. 1. Respuesta inmunológica e inflamatoria esquematizada, en la cual se demuestran las relaciones entre los diferentes componentes del síndrome de resistencia a la insulina y la aterosclerosis (véase texto). NF-*ß: factor nuclear kappa-beta; IL-1: interleucina 1; IL-6: interleucina 6; FNT*: factor de necrosis tumoral alfa; PCR: proteína C reactiva; PSAA: proteína sérica A-amiloide; Lp(a); lipoproteína A; PAI-1: inhibidor tipo 1 del activador del plasminógeno; LDL: lipoproteína de baja densidad; VLDL: lipoproteína de muy baja densidad; HDL: lipoproteína de alta densidad; ACTH: hormona corticotropa adrenal.
Inflamación, diabetes y desarrollo de la placa aterosclerótica
La etiopatogenia de la aterosclerosis es multifactorial, lo cual se verifica con claridad en el paciente diabético67. La disfunción endotelial está considerada como el estadio más precoz en el proceso aterogénico68 y se ha asociado a factores de riesgo cardiovascular clásicos y otros más recientes, como la hiperhomocisteinemia, el estrés oxidativo y las infecciones crónicas69. La consecuencia fisiológica de la disfunción endotelial es la disminución en la producción de óxido nítrico (ON), con la consiguiente pérdida de su función vasomotora y antiaterogénica (antiagregante, antiadhesiva, antiproliferativa y antioxidante). Además, la disfunción endotelial se asoció a un aumento de sustancias vasoconstrictoras, como las endotelinas70. El primer cambio histopatológico en el proceso aterogénico es la acumulación de LDL en el espacio subintimal y la subsecuente oxidación, llevada a cabo principalmente por macrófagos. Las LDL oxidadas son capaces de activar al endotelio suprayacente71-73. Las células endoteliales activadas sintetizan selectinas, moléculas de adhesión y sustancias quimioatractivas que facilitan la unión y la posterior migración de linfocitos T y monocitos circulantes al espacio subendotelial. Aquí, los monocitos maduran a macrófagos, internalizan las LDL oxidadas y se transforman en células espumosas, las cuales producen más radicales libres y liberan nuevas citocinas para la atracción de más macrófagos y células musculares lisas (CML)73,74.
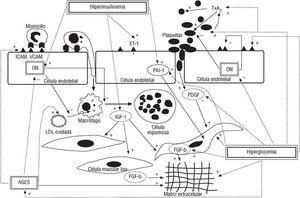
Se ha demostrado que personas jóvenes, familiares de primer grado de pacientes diabéticos, obesos o individuos insulinorresistentes, presentan disfunción endotelial con una vasodilatación dependiente del endotelio alterada, con independencia de otros factores de riesgo cardiovascular75,76. Experimentalmente, la insulina produce vasodilatación dependiente de ON, pero en presencia de hiperglucemia y de un estado de resistencia a la insulina dicha vasodilatación es abolida77. La hiperglucemia disminuye la disponibilidad de ON78 y aumenta la expresión de moléculas de adhesión en las células endoteliales79. Esto se debe a que la hiperglucemia induce un incremento en la producción de radicales libres y favorece la glicación no enzimática de proteínas y lipoproteínas, con la subsiguiente producción de productos avanzados de la glicación (AGE).Se ha detectado la acumulación, tanto de AGE como de proteínas glicoxidadas, en las placas de ateroma de pacientes diabéticos, lo que puede expresar la existencia de un mayor estrés oxidativo en la DM80,81. Los AGE promueven la expresión de moleculas de adhesión82, la inactivación de ON83, la migración de macrófagos84 y la oxidación de LDL glicadas en su interior85. Adicionalmente, la hiperglucemia disminuye la producción de prostaciclina, sustancia vasodilatadora y antiagregante86. Es interesante señalar que la hiperinsulinemia estimula la síntesis de la endotelina 1, potente sustancia vasoconstrictora87 (fig. 2). La hiperhomocisteinemia duplica el riesgo de muerte en los pacientes diabéticos, y se especula que actúa incrementando el ya alto grado de estrés oxidativo existente en la DM tipo 288.

Fig. 2. Efectos biológicos de la hiperglucemia, hiperinsulinemia y productos AGE sobre las distintas células implicadas en el proceso de la aterosclerosis. ON: óxido nítrico; ET-1: endotelina 1; PAI-1: inhibidor 1 del activador del plasminógeno; IGF-1: factor 1 de crecimiento insulina-like. PDGF: factor de crecimiento derivado de las plaquetas; FGF-ß: factor beta transformador del crecimiento; AGES: productos avanzados de la glicación; TxA2: tromboxano A2; ICAM: molécula 1 de adhesión intercelular; VCAM: molécula de adhesión de células vasculares; (+): efecto activador; (): efecto inhibidor.
Inflamación, diabetes y fisura de la placa aterosclerótica
La placa de ateroma es una estructura dinámica en la que existe un equilibrio entre la influencia destructiva de células inflamatorias y el efecto estabilizante de las células musculares lisas (CML)89. Hoy día sabemos que las placas más vulnerables son las que poseen un mayor núcleo lipídico, una capa fibrosa más fina y una mayor proporción de células inflamatorias90-93. Los linfocitos T activados elaboran interferón-* (IFN-*) que inhibe la proliferación de las CML y su capacidad de síntesis de colágeno. Los macrófagos producen metaloproteinasas que degradan las proteínas de la matriz extracelular, sintetizan factor tisular e inducen apoptosis de las CML. Estos efectos celulares producen el adelgazamiento de la placa fibrosa, lo que predispone a un mayor riesgo de rotura93. Cuando se produce la rotura o erosión de la placa de ateroma se exponen a la sangre el core lipídico con el factor tisular y la matriz colágena, que son altamente trombogénicos. Este acontecimiento induce la activación y la agregación plaquetaria, el depósito de fibrina y la formación del trombo94. Recientemente, Moreno et al95 demostraron que las placas ateromatosas de los pacientes diabéticos tienen un mayor contenido lipídico y una mayor infiltración macrofágica que las de los pacientes no diabéticos. También se sabe que las plaquetas de los pacientes diabéticos muestran una mayor adhesividad y agregabilidad96. Además, la actividad de la vía del ácido araquidónico está incrementada en los pacientes diabéticos, presentan una mayor síntesis de tromboxano A2, potente vasoconstrictor y activador plaquetario97. Adicionalmente, la hiperinsulinemia incrementa las concentraciones del PAI-1 tanto en la sangre como en la pared arterial de los pacientes diabéticos46,47. El PAI-1 inhibe la migración de las CML, lo que predispone a la formación de placas con una capa fibrosa fina y, por tanto, con una mayor predisposición a la rotura98 (fig. 2). Así mismo, en muestras de reestenosis coronaria de pacientes diabéticos se observó un gran componente colágeno.Estos hallazgos sugieren que en el paciente diabético la placa es muy trombogénica99.Esto explica la gran propensión a la rotura, trombosis y cicatrización, entrando en un círculo vicioso de dos procesos muy agresivos en la DM como son el crecimiento y la vulnerabilidad de la placa. La mayoría de estos episodios son clínicamente silentes, lo que explica que en el momento del diagnóstico de la DM el riesgo cardiovascular del paciente diabético es el mismo que el de una persona no diabética con antecedentes isquémicos.
Inflamación, diabetes y reestenosis
La DM se asocia a un mayor índice de reestenosis tras los procedimientos de revascularización percutánea100. La introducción del stent ha reducido su incidencia desde un 40 a un 15-20% según las series101. La reestenosis resulta de la compleja relación entre la trombosis, el remodelado vascular y la proliferación neointimal. La formación de la neoíntima es determinante en la reestenosis y es consecuencia de la acumulación de CML y de matriz extracelular102.Se ha demostrado que la insulina in vitro es un agente mitógeno de diversos tipos celulares, como las CML y los fibroblastos103. El estado de resistencia a la insulina y la hiperglucemia aumentan la síntesis de factores que estimulan la proliferación de CML, como el factor de crecimiento derivado de las plaquetas (PDGF) y el factor 1 de crecimiento insulina-like (IGF-1). Otros factores inducen la producción de matriz extracelular por las CML, como el factor beta transformador del crecimiento (FGF-ß)104 (fig. 2). Además, la hiperglucemia inhibe la síntesis de heparán sulfato de la matriz extracelular, que es un potente inhibidor de las CML, lo que facilita su proliferación100. Adicionalmente, los AGE ayudan al proceso de reestenosis al reclutar y activar células inflamatorias, y alterar las propiedades funcionales de los componentes de la matriz extracelular mediante el proceso de glicación105. El tratamiento de la reestenosis es un reto para la cardiología actual y no existe ninguna terapia efectiva en la prevención de la hiperplasia intimal, a excepción de la braquiterapia106, que parece ofrecer resultados prometedores.
Resistencia a la insulina, hiperinsulinemia y factor nuclear-*ß
Actualmente conocemos que muchos de los genes que codifican proteínas clave en la respuesta inflamatoria de la aterosclerosis son modulados por el factor de transcripción nuclear-*ß (NF-*ß) (tabla 1). El NF-*ß reside de forma inactiva en el citoplasma de diversos tipos celulares y se activa por numerosos estímulos como el estrés oxidativo, las citocinas, diversos mitógenos, etc.107. Su activación induce la expresión de interleucinas, TNF-*, INF-*, moléculas de adhesión, metaloproteinasas y otros factores proinflamatorios clave en el desarrollo y la progresión de la aterosclerosis107.Sabemos que el aumento de la actividad de este factor de transcripción nuclear se correlaciona con la extensión y severidad de la enfermedad coronaria (García-Moll et al, datos no publicados) y con la actividad de la placa108. Recientemente se ha demostrado de manera experimental que la insulina per se no activa el NF-*ß, pero potencia el efecto de la hiperglucemia, los AGE y la angiotensina II en la activación del NF-*ß, lo que ha llevado a la hipótesis de que en el microambiente resultante de la resistencia a la insulina existen múltiples factores que pueden activarlo y, en consecuencia, acelerar el proceso de la aterosclerosis109.

DIABETES Y ATEROSCLEROSIS. PAPEL DE LOS RECEPTORES GAMMA ACTIVADOS DEL PEROXISOMA PROLIFERADOR (PPAR-*)
Resistencia a la insulina, aterosclerosis y PPAR-*
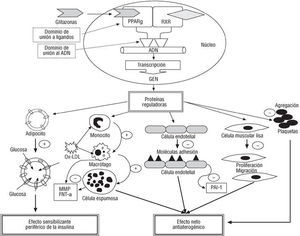
El receptor gamma activado del peroxisoma proliferador (PPAR*) es un receptor nuclear que actúa como factor de transcripción dependiente del ligando (fig. 3)110.El PPAR* se expresa en los tejidos diana de la insulina y su activación regula el metabolismo de la glucosa y de los ácidos grasos111. Agonistas de los PPAR*, como los nuevos antidiabéticos orales de la clase de las glitazonas, han demostrado aumentar
la sensibilidad periférica a la insulina y mejorar el estado de resistencia a la insulina en pacientes diabéticos tipo 2112. Además del control glucémico, los pacientes diabéticos mejoraron todos los componentes del síndrome metabólico (disminución de triglicéridos, ácidos grasos, LDL, PAI-1, cifras tensionales y aumento de las HDL)113,114 (tabla 2) y en un estudio se demostró regresión de la aterosclerosis115. El reciente descubrimiento de los PPAR* nos ofrece otra evidencia más de la asociación entre la resistencia a la insulina, la DM y la aterosclerosis.

Fig. 3. Representación esquemática del receptor * activado del peroxisoma proliferador y del receptor X retinoide, que actúan conjuntamente. En el núcleo de diversos tipos celulares regulan la transcripción de proteínas clave en el desarrollo del proceso de la aterosclerosis y de la resistencia a la insulina (véase texto). PPAR*: receptor * activado del peroxisoma proliferador; RXR: receptor X retinoide; Ox-LDL: lipoproteínas de baja densidad oxidadas; MMP: metaloproteinasas; FNT-*: factor de necrosis tumoral alfa; PAI-1: inhibidor 1 del activador del plasminógeno.

Inflamación, aterosclerosis y PPAR*
Los PPAR* se expresan en células presentes dentro de la placas de ateroma humanas116. Experimentalmente, en células humanas los agonistas del PPAR* inhiben la expresión de moléculas de adhesión, endotelinas y PAI-1 en células endoteliales117-119. También inhiben la proliferación de las CML120. En el macrófago inhiben la liberación de metaloproteinasas121, producen su apoptosis y regulan su metabolismo lipídico122. Así mismo, estimulan en el macrófago la expresión de otro tipo de receptores (CLA-1/SR-BI) que se unen con gran afinidad a las HDL y desempeñan un papel importante en el transporte del colesterol desde los vasos sanguíneos al hígado. Por otro lado, se han descrito acciones proaterogénicas en el macrófago; así, la activación de los PPAR* promueve la internalización de LDL oxidadas y la transformación del macrófago en célula espumosa. Recientemente se ha demostrado que el estado de hiperglucemia en la DM tipo 2 produce una alteración en la regulación de los PPAR* de los macrófagos, lo que probablemente es determinante en la severidad y prematuridad de la aterosclerosis en los pacientes diabéticos123. Como vemos, la activación de los PPAR* regula la homeostasis del colesterol y de la placa ateromatosa en distintas localizaciones con acciones tanto proinflamatorias como antiinflamatorias, pero el efecto neto es antiaterogénico.
En pacientes diabéticos tipo 2, el tratamiento con glitazonas, además de sus efectos metabólicos, mejoró la disfunción endotelial y los episodios de angina vasospástica124, redujo la proliferación neointimal tras la implantación de stent coronario125, mejoró la función sistólica del ventrículo izquierdo126 y redujo las concentraciones de reactantes de fase aguda127, LDL oxidadas y moléculas de adhesión128. Por tanto, se abre una nueva y esperanzadora vía de actuación en el complejo mundo de la resistencia a la insulina, DM y enfermedad coronaria. Estudios futuros, tanto en pacientes diabéticos como en no diabéticos, nos aportarán información sobre si el efecto antiaterogénico se debe a una acción vascular directa, a la mejoría en las alteraciones metabólicas, o a ambos efectos.
TERAPIA ANTIINFLAMATORIA, ATEROGÉNESIS Y DIABETES
La tendencia actual es la implantación de un esquema terapéutico multifactorial y agresivo en el paciente diabético. Con la adquisición de buenos hábitos higiénico-dietéticos podemos mejorar el estado de la resistencia a la insulina y el control de los factores de riesgo asociados. Cuando estas medidas no son suficientes debemos implantar la terapia farmacológica más adecuada. Así, tanto en prevención primaria como en secundaria disponemos de estatinas y fibratos para el tratamiento de la dislipemia aterogénica, que es determinante en la afectación macrovascular. Se ha demostrado que el beneficio de las estatinas no se explica por completo por el efecto antilipídico. Recientemente, la pravastatina demostró reducir las concentraciones de PCR con independencia de la magnitud en la reducción lipídica en pacientes que habían presentado un infarto de miocardio129. Diversas estatinas han demostrado mejorar la disfunción endotelial, estabilizar la placa y tener un efecto antitrombótico130. Tanto las estatinas como los fibratos son agonistas del PPAR* y pueden tener, por tanto, un efecto antiinflamatorio. En un reciente estudio, los fibratos han demostrado reducir las concentraciones de IGF-1, junto con un enlentecimiento en la progresión de la aterosclerosis131. Como tratamiento antitrombótico, el uso de salicilatos está establecido en prevención secundaria y la Asociación Americana de la Diabetes recomienda su uso en prevención primaria en pacientes diabéticos, sobre todo en aquellos de más alto riesgo132. Además de la inhibición de la enzima ciclooxigenasa, estos agentes pueden tener mecanismos adicionales, incluida la inhibición del NF-*ß. El uso de salicilatos demostró disminuir el riesgo de infarto de miocardio en varones sanos y se asoció de manera significativa con una reducción en las concentraciones plasmáticas de la PCR133. En otro trabajo, el ácido acetilsalicílico disminuyó la concentración de citocinas proinflamatorias y la PCR en pacientes con enfermedad coronaria documentada134. Tampoco debemos olvidar el efecto beneficioso de los antagonistas de los receptores IIb/IIIa plaquetarios en pacientes diabéticos con síndromes coronarios agudos con o sin intervencionismo coronario135,136. Otras medidas, como el uso de antioxidantes, pueden resultar especialmente útiles en el paciente diabético, en el cual los procesos oxidativos están incrementados137. Las nuevas glitazonas, como hemos comentado con anterioridad, mediante sus acciones antiinflamatoria y sensibilizante periférica de la insulina, son fármacos prometedores en el tratamiento del enfermo diabético. Además de la utilización de estos agentes con acciones antiinflamatorias y sensibilizantes a la insulina, es imprescindible insistir en la importancia de un buen control de las alteraciones metabólicas de la DM. Aunque existe controversia en cuanto al papel de la hiperglucemia en el desarrollo de la enfermedad coronaria, el estudio Diabetes and Insulin-Glucose Infusion in Acute Myocardial Infarction (DIGAMI)12 tras infarto de miocardio ha demostrado que el buen control glucémico disminuye significativamente la mortalidad a largo plazo. En el campo de la prevención primaria, el estudio United Kingdom Prospective Diabetes Study (UKPDS)50 demostró que el buen control metabólico reducía en un 16% la incidencia de infarto de miocardio. En otro reciente estudio se halló que, tras efectuar una angioplastia138, el principal determinante de mal pronóstico a largo plazo era el mal control glucémico, y no necesariamente el tamaño del vaso.
Por tanto, concluimos que la DM se asocia de forma compleja, pero directa, con una temprana y rápida progresión de la aterosclerosis coronaria. Los mecanismos inmunitarios e inflamatorios que subyacen al proceso aterogénico y que parecen influir de manera directa en el crecimiento y la rotura de la placa de ateroma son particularmente agresivos en el paciente diabético. La implantación de un plan terapéutico temprano y racional, tanto en prevención primaria como en secundaria, puede reducir el incrementado riesgo cardiovascular que presentan los pacientes diabéticos. Nuevos antidiabéticos orales con propiedades antiinflamatorias y sensibilizantes a la acción de la insulina, que actúan modificando la transcripción genética de factores clave implicados en el proceso de la aterosclerosis, han abierto una nueva vía de actuación terapéutica en el tratamiento del paciente diabético.
El Dr. A. Sánchez-Recalde recibe una beca de la Sociedad Española de Cardiología. Correspondencia: Prof. J.C. Kaski. Head of Coronary Artery Disease Research Unit. Department of Cardiological Sciences. St. George's Hospital Medical School. London SW17 ORE. Correo electrónico: jkaski@sghmd.ac.uk