La insuficiencia cardiaca aguda (ICA) se ha definido como la aparición o el agravamiento de signos y síntomas de insuficiencia cardiaca (IC) que requiera un tratamiento urgente1. La ICA es una de las principales causas de morbilidad y mortalidad2. A pesar de la considerable variación existente en los perfiles clínicos y la heterogeneidad sustancial de las causas subyacentes, la inmensa mayoría de los pacientes con ICA presentan signos y síntomas de congestión pulmonar y sistémica más que de bajo gasto cardiaco. En consecuencia, la disnea es el síntoma de presentación cardinal en los pacientes hospitalizados por ICA3.
Aunque muchos pacientes responden al tratamiento inicial, hay un porcentaje significativo que no muestra un alivio rápido de la disnea1. Además, hay una disociación entre la presión capilar pulmonar enclavada (PCPE) y la gravedad de la disnea, de modo que pacientes con una PCPE elevada pueden presentar una disnea mínima, mientras que otros con una PCPE comparativamente inferior pueden sufrir disnea grave4. Asimismo, la tasa de mortalidad y de reingresos a corto plazo es de hasta un 50%5. Estas observaciones resaltan el conocimiento incompleto de que disponemos sobre la patogenia de la congestión pulmonar en la ICA.
FISIOPATOLOGÍA DE LA CONGESTIÓN PULMONARLa congestión pulmonar se define como la acumulación de líquido en los pulmones, que da lugar a deterioro del intercambio gaseoso e hipoxemia arterial. Se produce secuencialmente: aparece primero en la región hiliar de los pulmones, luego llena el espacio intersticial y, finalmente, en su forma más grave, inunda los alveolos. La presión de llenado del ventrículo izquierdo (VI) elevada, que conduce a hipertensión venosa pulmonar (aumento de la PCPE) es el principal mecanismo subyacente en la congestión pulmonar. La elevación de la presión diastólica del VI (PDVI) es consecuencia de la sobrecarga de líquidos causada por su retención o su redistribución6. Por otro lado, un aumento rápido de la presión arterial (poscarga), en especial en pacientes con disfunción diastólica, puede desencadenar una congestión pulmonar grave7. A menudo, la elevación de la PDVI (congestión hemodinámica) precede a la congestión clínica en días o incluso semanas8.
VIEJOS Y NUEVOS CONCEPTOS EN LA PATOGENIA DEL EDEMA PULMONAREl edema pulmonar es el resultado de un desequilibrio entre las fuerzas que ocasionan la entrada de líquido en los alveolos y los mecanismos para retirarlo. La filtración de líquido a través de la pared de los capilares pulmonares se describe con la ecuación de Starling9:

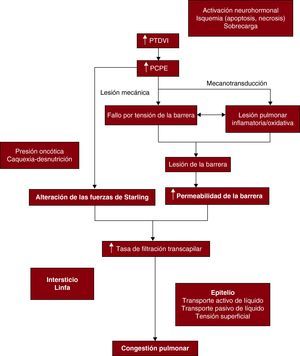
Según la ecuación de Starling, el equilibrio entre las presiones hidrostáticas (Pc – Pi) y las presiones oncóticas (πc – πi) constituye la fuerza que impulsa la filtración de líquido. Partiendo de este modelo simplista, el edema pulmonar se ha clasificado tradicionalmente en las categorías de cardiogénico y no cardiogénico. El edema pulmonar cardiogénico o hidrostático se debe a unas presiones hidrostáticas capilares pulmonares elevadas, que alteran el equilibrio de Starling mientras que la barrera alveolocapilar se mantiene intacta. Por el contrario, el edema no cardiogénico o de alta permeabilidad se caracteriza por la lesión de la barrera alveolocapilar con una fuga de líquido rico en proteínas hacia el intersticio y los espacios aéreos10. Sin embargo, este modelo fisiopatológico del movimiento pasivo de líquido, que depende de los gradientes oncótico e hidrostático de un lado a otro de la barrera sangre-gas, parece ser una simplificación excesiva. Los estudios basados en el cociente de proteínas del líquido de edema respecto a las proteínas del suero en los pacientes con edema pulmonar cardiogénico y no cardiogénico han puesto de manifiesto que con frecuencia hay una combinación de una presión capilar pulmonar hidrostática elevada y una permeabilidad elevada de la barrera alveolocapilar, que resulta en un notable solapamiento entre los dos grupos. Si el aumento de la presión capilar pulmonar hidrostática fuera de por sí la causa de la formación del edema pulmonar, sería previsible que la concentración de proteínas en el líquido del recubrimiento alveolar se redujera como consecuencia de la entrada de ultrafiltrado del plasma. Paradójicamente, esta concentración aumenta a casi el doble11, 12. Así pues, el edema pulmonar hidrostático o de alta permeabilidad puede corresponder a los extremos del espectro del edema pulmonar11, 12. Hay dos procesos fundamentales que pueden conducir a la disfunción de la barrera alveolocapilar en la ICA: a) la lesión mecánica de la barrera a causa del aumento de las presiones capilares pulmonares hidrostáticas, y b) la lesión pulmonar inflamatoria y oxidativa (Figura 1).

Figura 1. Diagrama en el que se muestra la intervención de la lesión mecánica y la lesión pulmonar inflamatoria y oxidativa en la disfunción de la barrera alveolocapilar y la congestión pulmonar en pacientes con insuficiencia cardiaca aguda. PCPE: presión capilar pulmonar enclavada; PTDVI: presión telediastólica ventricular izquierda.
PROPIEDADES FISIOLÓGICAS DE LA BARRERA ALVEOLOCAPILAREn sus zonas más delgadas, la barrera sangre-gas está formada por la capa de endotelio capilar, la capa de epitelio alveolar y la matriz extracelular, que se origina por la fusión de las membranas basales de estas dos capas13, 14. La barrera sangre-gas en el pulmón humano debe desempeñar dos funciones contradictorias. Por un lado, debe ser extremadamente delgada para fomentar un intercambio eficiente del oxígeno y el dióxido de carbono mediante difusión pasiva y, por otro, tiene que ser lo suficientemente fuerte para superar la tensión que le impone la presión hidrostática capilar elevada. La pérdida de su integridad estructural puede dar lugar a edema o hemorragia alveolar. La resistencia de la barrera sangre-gas puede atribuirse al tipo de colágeno de las membranas basales15.
DISFUNCIÓN AGUDA Y CRÓNICA DE LA BARRERA SANGRE-GAS EN LA INSUFICIENCIA CARDIACASe ha introducido el término «fallo por tensión» para describir la lesión mecánica de la barrera alveolocapilar que se produce como resultado de un aumento brusco de la presión hidrostática capilar pulmonar16. En varios modelos experimentales se ha observado que el traumatismo inducido por la presión conduce a alteraciones ultraestructurales de la barrera sangre-gas que comportan la rotura de la capa de endotelio capilar pulmonar, así como de la capa de epitelio alveolar16. El resultado es una transición progresiva de una forma de edema pulmonar de baja permeabilidad a otra de alta permeabilidad17. Hay evidencias experimentales de que las alteraciones ultraestructurales de la barrera sangre-gas que se observan en la lesión mecánica aguda son reversibles18. Por otra parte, la elevación persistente de la presión capilar pulmonar conduce a un engrosamiento de la barrera alveolocapilar a causa del depósito excesivo de colágeno tipo IV15. Este proceso de remodelado puede tener efectos protectores contra un mayor daño por presión elevada y puede aumentar la resistencia del pulmón al desarrollo de edema pulmonar en los pacientes con IC crónica11. Sin embargo, causa una reducción significativa de la capacidad de difusión alveolar y deteriora la transferencia de gases y la capacidad de ejercicio. Las proteínas específicas del epitelio pulmonar pueden atravesar la barrera alveolocapilar y pasar a la circulación, y serían marcadores de una lesión de la barrera en varios trastornos patológicos19. La proteína B surfactante (SP-B) es la proteína específica surfactante más pequeña que se puede detectar en la circulación. La SP-B desempeña un papel crucial en la formación y la estabilización del surfactante pulmonar; se sintetiza exclusivamente por las células del epitelio alveolar tipo II que la secretan a través de su superficie apical hacia el interior de los alvéolos, de modo que, en circunstancias normales, se mantiene un gradiente de líquido de recubrimiento epitelial:plasma > 1.500:120. Sin embargo, en el caso de que la barrera esté dañada, se produce una fuga de mayores cantidades hacia el torrente circulatorio. Así pues, la cantidad de SP-B circulante aumenta de manera aguda en respuesta a la disfunción del VI inducida por el ejercicio, probablemente debido a una disfunción de la barrera que es consecuencia de un aumento agudo de las presiones hidrostáticas capilares pulmonares21. Además, se ha descrito un aumento prolongado de la SP-B circulante tras el edema pulmonar cardiogénico agudo, lo cual indica una lesión persistente de la barrera en esos pacientes22. Por último, la concentración plasmática circulante de SP-B está relacionada con la difusión gaseosa alveolar, el rendimiento general en el ejercicio y la eficiencia de la ventilación, lo cual pone de manifiesto una relación entre la lesión anatómica y funcional de la barrera alveolocapilar en los pacientes con IC23.
Papel de la lesión pulmonar inflamatoria y oxidativa en el contexto de la insuficiencia cardiaca agudaLa lesión inflamatoria grave del endotelio capilar pulmonar y del epitelio alveolar, que lleva a una disfunción de la barrera y a la formación de un edema pulmonar de alta permeabilidad, desempeña un papel clave en la fisiopatología de la lesión pulmonar aguda y de su manifestación más grave, el síndrome de dificultad respiratoria aguda (SDRA). Sin embargo, cada vez hay más evidencia de que la lesión pulmonar hidrostática en el contexto de la ICA está relacionada con la inflamación pulmonar24. El líquido del edema pulmonar en la ICA presenta un aumento de neutrófilos25, citocinas proinflamatorias26 y biomarcadores de estrés oxidativo. Además, la disfunción prolongada de la barrera sangre-gas tras un edema pulmonar cardiogénico agudo puede estar relacionada con la inflamación del parénquima pulmonar22.
La inflamación del pulmón puede formar parte del mecanismo de reparación tras la lesión hidrostática pulmonar. Como se ha señalado antes, el «fallo por tensión» de la barrera sangre-gas puede conducir a una transición progresiva de la forma de edema pulmonar de baja permeabilidad a la de alta permeabilidad. La eliminación por los macrófagos de las proteínas precipitadas en los alveolos durante la resolución del edema pulmonar puede producir una actividad inflamatoria como la liberación de factor de necrosis tumoral alfa27, 28.
Por otra parte, la inflamación del pulmón en el contexto de la ICA puede ser una respuesta directa a la tensión mecánica de la microcirculación pulmonar. En el epitelio pulmonar puede producirse una transducción de la señal mecánica a una respuesta biológica, mediante la inducción de varias vías de señalización intracelular, que pueden comportar un aumento de la producción de citocinas inflamatorias, activación de los macrófagos, inflamación aguda y disfunción de la barrera29. De entre las diversas vías de señalización inducidas por la tensión mecánica de la microcirculación pulmonar, cada vez se presta mayor atención al papel de las especies moleculares de oxígeno reactivo. El estrés oxidativo desempeña un papel importante en el deterioro de la barrera sangre-gas, ya sea por una lesión oxidativa directa de componentes celulares básicos de la barrera o a través de la activación de vías de señalización sensibles a la actividad redox que conduce a la apoptosis y la inflamación29.
La lesión pulmonar inflamatoria y oxidativa puede desempeñar un papel fisiopatológico importante en la descompensación de la IC al dañar en mayor medida la barrera alveolocapilar y aumentar su permeabilidad. Como consecuencia de ello, el umbral de presión hidrostática capilar pulmonar para la acumulación de líquido pulmonar se reduce. Este parámetro podría explicar la vulnerabilidad de los pacientes con ICA a las recurrencias.
EVALUACIÓN DE LA LESIÓN PULMONAR EN LA INSUFICIENCIA CARDIACA AGUDAEl examen del líquido del recubrimiento epitelial puede aportar información útil acerca del daño sufrido por la barrera alveolocapilar en pacientes con IC, sobre todo en lo relativo a procesos fisiopatológicos importantes como la inflamación y la alteración redox. Hasta el momento, el acceso a este líquido se ha basado en el lavado broncoalveolar, una técnica invasiva que requiere broncoscopia y puede influir en el grado de inflamación de las vías aéreas. Por consiguiente, se dispone de escasa información procedente de un reducido número de pacientes con edema pulmonar cardiogénico y necesidad de ventilación mecánica26, 30. Recientemente ha aumentado el interés por la toma de muestras de vías respiratorias bajas con técnicas no invasivas, como la inducción de esputo, la determinación del óxido nítrico espirado y la obtención y el análisis de condensado de aire espirado (CAE).
El CAE ha surgido como posible instrumento para el estudio del líquido del recubrimiento del epitelio alveolar. Está formado principalmente por agua con pequeñas gotas aerosolizadas atrapadas que proceden del líquido del recubrimiento de las vías aéreas, así como compuestos hidrosolubles volátiles y no volátiles31. Su principal componente es vapor de agua condensado, que constituye casi la totalidad del volumen (> 99%) de líquido recogido en el CAE32. La obtención de CAE es una técnica sencilla, completamente no invasiva, segura y reproducible. Se realiza mediante la espiración del volumen corriente en un aparato de condensación con enfriamiento. Se ha estudiado una amplia gama de biomarcadores en el CAE, como pH, citocinas, isoprostanos, leucotrienos, óxidos de nitrógeno, péptidos, adenosina, metabolitos del ácido araquidónico, amonio, peróxido de hidrógeno y ADN33. Así pues, el CAE se ha venido utilizando de manera creciente como instrumento de investigación y herramienta clínica en el estudio de la inflamación de vías aéreas, el estrés oxidativo y el equilibrio acidobásico en muchas enfermedades pulmonares, como asma, enfermedad pulmonar obstructiva crónica, SDRA, fibrosis quística, bronquiectasias y cáncer de pulmón34. Para aportar nuevas perspectivas sobre el papel de la lesión pulmonar y la disfunción de la barrera sangre-gas en la ICA, hemos demostrado que, durante los episodios de descompensación de la IC, el CAE muestra un aumento de los marcadores de actividad inflamatoria y estrés oxidativo (datos no publicados).
En conclusión, la congestión pulmonar en la ICA es un proceso fisiopatológico complejo, que va más allá de la sobrecarga de líquido y la hemodinámica. La lesión pulmonar inflamatoria y oxidativa que causa una disfunción de la barrera sangre-gas parece ser clave en la patogenia del edema pulmonar y puede constituir una nueva diana terapéutica. Serán necesarios nuevos estudios para aclarar si la prevención de la lesión de la barrera, en vez de controlar simplemente las presiones capilares pulmonares hidrostáticas, permite mejorar el tratamiento y el pronóstico de la ICA.
CONFLICTO DE INTERESESNinguno.
Full English text available from: www.revespcardiol.org
Autor para correspondencia: Heart Failure Unit, Athens University Hospital, 12461 Atenas, Grecia. geros@otenet.gr