
De acuerdo con la filosofía acerca de lasguías de práctica clínica marcada por el Comité Ejecutivo de la SEC (Sociedad Española de Car-diología)1, se ha redactado un documento que pretende debatir los aspectos más importantes y novedosos de la guía sobre el diagnóstico y manejo de la miocardiopatía hipertrófica (MCH), elaborada por la Sociedad Europea de Cardiología (ESC)2.
Las guías actuales sustituyen en el ámbito europeo al documento de consenso del American College of Cardiology (ACC) y la ESC publicado en 20033 y que en Norteamérica había sido reemplazado en 2011 por la guía de práctica clínica del ACC y la American Heart Association (AHA)4. La presente guía es muy pertinente dada la necesidad de actualizar el conocimiento recogido en el documento de 2003, pero también debido a diversas y relevantes discrepancias que existen en la práctica cardiológica europea con respecto a lo recogido en las guías ACC/AHA de 20114.
MÉTODOSEl Comité de Guías de Práctica Clínica de la SEC ha creado un grupo de trabajo integrado por cardiólogos expertos en diferentes áreas, propuestos por las Secciones de Cardiología Clínica e Insuficiencia Cardiaca y Trasplante, así como por el Grupo de Trabajo de Cardiopatías Familiares de la SEC. El objetivo general de este grupo de trabajo era revisar las evidencias y recomendaciones aportadas por la guía europea sobre diagnóstico y manejo de la MCH, que es la aceptada por la SEC, y cuya traducción aparece publicada en este número de REVISTA ESPAÑOLA DE CARDIOLOGÍA. A todos los integrantes del grupo de trabajo se les solicitó que realizaran un análisis de la guía. Específicamente se les solicitó que comentasen: a) aportaciones novedosas y más trascen-dentes para la práctica clínica; b) aspectos más positivos y más conflictivos de esas aportaciones novedosas; c) implicaciones para la práctica clínica en nuestro país y en nuestro entorno. Con esta infor-mación se redactó un documento que recoge todas las aportaciones de los participantes. Este artículo fue revisado por 14 expertos evalua-dores externos, y tras una definitiva revisión por el grupo de trabajo, se envió a la revista para su evaluación y eventual publicación simul-táneamente con la versión española de la guía europea.
COMENTARIOS GENERALES Y ANÁLISIS DE LA METODOLOGÍALa MCH es la enfermedad genética cardiaca más común y la causa más frecuente de muerte súbita (MS) en jóvenes2–4. No hay datos relativos a la prevalencia de la MCH en España, pero múltiples trabajos realizados en diversas poblaciones han evidenciado que la prevalencia de la MCH es de 1 cada 500 personas de la población general2–4.
Si extrapolamos estos datos de prevalencia, en España existen más de 90.000 personas con MCH. Por tanto, esta guía es particularmente útil, atendiendo al importante número de pacientes que padecen MCH en nuestro país y a las particularidades del manejo de diversas compli-caciones de esta enfermedad (obstrucción del tracto de salida del ven-trículo izquierdo, embolia o riesgo de MS). Además, los avances de la última década en el campo de la genética y su irrupción en la práctica asistencial, también hacían que fuera muy conveniente un marco de recomendaciones para el cardiólogo general. Pese a que en España existen unidades de cardiopatías familiares y unidades de referencia desig-nadas por el Ministerio de Sanidad para la atención de los pacientes con MCH5, la mayoría de los pacientes son atendidos fuera de ellas. Por tanto, una guía general actualizada sobre diagnóstico y manejo de la MCH es muy bien recibida. A pesar de que la guía está concebida para su adopción por un cardiólogo no especialista en la enfermedad, en numerosas ocasiones se hace referencia a la conveniencia de que diversos aspectos de la atención a estos enfermos (estudios genéticos, con-sejo familiar y reproductivo, manejo invasivo de la obstrucción, etc.) se realicen en unidades de referencia con experiencia contrastada.
La guía en su contenido es bastante detallada y extensa (55 pági-nas, con 506 citas bibliográficas y 36 tablas de recomendaciones).
La metodología global es similar a la de otras guías de la ESC: tras una descripción del estado actual de cada tema, en una serie de tablas se indican las recomendaciones en ese tema (I, IIa, IIb o III) y el peso de la evidencia (A, B o C) que apoya dichas recomendaciones. Dada la falta de ensayos clínicos en la MCH, no es de extrañar que de las 132 recomendaciones de la guía, 96 (73%) son recomendaciones con un nivel de evidencia C, es decir, recomendaciones basadas en registros, estudios retrospectivos o consenso de expertos. El 27%, 36, están apoyadas en una evidencia B (derivada de un solo ensayo aleatorizado o de múltiples estudios no aleatorizados) y ninguna recomendación presenta un nivel de evidencia A (basada en múltiples estudios alea-torizados y metanálisis).
Esta situación debería hacer reflexionar sobre la necesidad de rea-lizar ensayos clínicos diseñados para dar una respuesta científica a muchas cuestiones relacionadas con esta enfermedad y que actual-mente se resuelven mediante el consenso de expertos.
VALORACIÓN DE LOS ASPECTOS MÁS RELEVANTESLos aspectos más relevantes o novedosos identificados por el grupo de trabajo son los siguientes: diagnóstico etiológico; criterios diagnósticos; evaluación diagnóstica; pruebas genéticas y cribado familiar; manejo de la obstrucción; manejo de síntomas en ausencia de obstrucción; arritmias auriculares; prevención de la MS; reproduc-ción y contracepción, y situaciones especiales.
Diagnóstico etiológicoEl aspecto más novedoso en este apartado es que se hace una aproximación detallada a las diversas causas de la enfermedad.
En línea con la clasificación de las miocardiopatías de la ESC, la clasificación de la MCH está basada en criterios morfológicos, y se agrupan las etiologías en familiares/con base genética y no familiares/sin base genética.
En el documento se resalta que en la mayoría de los pacientes la enfermedad tiene una base genética, pudiéndose agrupar en 3 grupos:
- 1.
Mutaciones en genes de proteínas sarcoméricas (40-60%), donde las mutaciones en MYBPC3 y MYH7 son las más frecuentes.
- 2.
Mutaciones en otros genes no sarcoméricos (5-10%), donde se incluirían diferentes trastornos metabólicos (como la enfermedad de Fabry, la enfermedad de Danon, etc.), enfermedades neuromus-culares, amiloidosis familiar relacionada con TTR (relacionada con transtiretina) y síndromes malformativos (síndrome de Noonan, síndrome de LEOPARD, etc.).
- 3.
Otras causas sin base genética, como algunas amiloidosis y trans-tornos endocrinos.
En las tablas del anexo 2 se hace una descripción muy detallada de los genes implicados en la MCH. Con todo existe entre un 25-30% de enfermos en los que no se encuentra el defecto genético causal de la enfermedad, en los que la enfermedad puede deberse a mutaciones en nuevos genes todavía no descritos.
Aunque se hace una revisión breve de las distintas etiologías, la guía no proporciona pautas de tratamiento en las que disponen de tratamiento específico (p. ej., enfermedad de Fabry, amiloidosis fami-liar).
Cabe destacar que, tanto en este apartado como en otros, se lleva a cabo una exposición detallada de signos, síntomas y características particulares que permiten al clínico orientar el proceso diagnóstico etiológico. Así, se enumeran diversas características que deben inves-tigarse en todo paciente con MCH con el fin de identificar potenciales fenocopias de la enfermedad (tabla 1).
Signos y síntomas específicos para el diagnóstico etiológico en la miocardiopatía hipertrófica
| Síntomas | • Acroparestesias, tinnitus, sordera (Anderson-Fabry) debilidad muscular (enfermedades mitocondriales, Danon, FHL1) |
| Signos | • Retinitis pigmentosa (enfermedad de Danon, mitocondriales)• Córnea verticillata (Anderson-Fabry)Hipotensión postural (amiloidosis)• Síndrome del túnel carpiano (amiloidosis)Angiokeratoma, hipohidrosis (Anderson-Fabry)• Léntigos(LEOPARD)• Fenotipo facial (Anderson-Fabry, Noonan) |
| Electrocardiograma | • Preexcitación (PRKAG2, Danon, mitocondriales)• PR corto (Anderson-Fabry)• Bloqueo auriculoventricular (desminopatía, PRKAG2, Anderson-Fabry, amiloidosis, mitocondrial)• Bajos voltajes, patrón de seudoinfarto (amiloidosis) |
| Ecocardiografía | • Afectación concéntrica, biventricular (enfermedades infiltrativas o metabólicas)• Engrosamiento valvular (amiloidosis, Anderson-Fabry) |
| Historia familiar | • Diabetes, epilepsia, sordera (mitocondrial)• Ligado al X (Anderson-Fabry, Danon, FHL1)• Herencia materna (mitocondrial) |
| Bioquímica | • Elevación de creatincinasa (mitocondrial, Danon, FHL1)• Elevación ALT y AST (Danon)• Elevación de lactato (mitocondrial)• Insuficiencia renal (amiloidosis, Anderson Fabry, mitocondrial)• Paraproteína (amiloidosis AL) |
| Ergometría | • Acidosis grave prematura (mitocondrial) |
La presente guía clarifica la definición de la enfermedad para pacientes adultos y pediátricos. En adultos establece el punto diag-nóstico en un grosor ≥ 15mm en ausencia de causa que lo justifique y en niños lo sitúa en ≥ 2 desviaciones estándar con respecto a la media de grosor para su edad.
Para el diagnóstico de familiares es relevante la simplificación adoptada en la definición de la enfermedad. Se abandonan los criterios diagnósticos mayores y menores para familiares6 y se considera que un grosor ≥ 13mm es diagnóstico de MCH cuando se trate de un familiar de primer grado de un enfermo con MCH.
Aunque otros signos sutiles de las pruebas de imagen (criptas en resonancia magnética, etc.) o en el electrocardiograma pueden orien-tar a que un familiar está afectado, la simplificación propuesta ayudará en la estandarización de los familiares que se consideran afecta-dos o no afectados.
Evaluación diagnósticaLa guía hace una evaluación detallada de las diversas pruebas empleadas en la valoración de los pacientes con MCH, establece la utilidad de estas pruebas y con qué periodicidad deben realizarse.
A continuación se comentan algunos aspectos destacables en relación con estas pruebas.
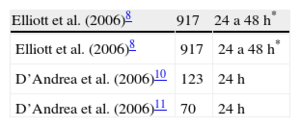
Holter-electrocardiogramaSe recomienda que la monitorización Holter-electrocardiograma sea de 48h. En nuestro país, la mayoría de centros dispone solo de monitorización de 24h, lo cual obligaría a realizar la prueba en 2 días distintos. Aunque hay algunos trabajos clásicos en los que el periodo de grabación de Holter-electrocardiograma ha sido mayor de 24 h7,8 es importante destacar que el Holter de 24h sigue siendo válido. Es más, la ecuación para el cálculo del riesgo de MS propuesta en la guía se ha obtenido a partir del Holter de 24h en la mayoría de casos9 y un aná-lisis detallado de los estudios en los que se ha relacionado la presencia de taquicardia ventricular no sostenida con MS revela que en muchos de ellos se realizaron grabaciones<48h (tabla 2)7,8,10,11.
Duración del periodo de monitorización de Holter-electrocardiograma en los estudios que han relacionado la presencia de taquicadia ventricular no sostenida y muerte súbita en pacientes con miocardiopatía hipertrófica
| Elliott et al. (2006)8 | 917 | 24 a 48h* |
| Elliott et al. (2006)8 | 917 | 24 a 48h* |
| D’Andrea et al. (2006)10 | 123 | 24h |
| D’Andrea et al. (2006)11 | 70 | 24h |
ECG: electrocardiograma.
*La mayoría Holter de 48h.
Este es un aspecto controvertido, porque en la estratificación de riesgo de MS se utiliza el gradiente obtenido en reposo o Valsalva, pero no el obtenido con el ejercicio, cuando no es más que una manera (y más fisiológica) de provocar el gradiente.
En las guías previas2 no se otorgaba tanta importancia al gradiente en la estratificación de riesgo de MS.
Tamaño de la aurícula izquierdaLa guía recomienda medir el diámetro anteroposterior de la aurícula izquierda (AI). Es conocido que existe una gran variabilidad interoperador, e incluso intraoperador, en la medida del diámetro de la AI. Muchos centros en su práctica habitual, y no solo en pacientes con MCH, utilizan el volumen de la AI y obvian esta medida. Como en la estratificación de riesgo de MS se ha incluido el diámetro antero-posterior debe recomendarse encarecidamente que la medición sea lo más exacta posible.
Realce tardío en la resonancia magnéticaLa utilidad del realce tardío de gadolinio como marcador de riesgo de MS es un aspecto controvertido y una de las discrepancias más importantes con respecto a la guía ACC/AHA4. La guía estadounidense establecía que el realce extenso es un factor que influía en el implante de desfibrilador automático implantable (DAI) en pacientes que pre-sentan taquicardia ventricular no sostenida o respuesta presora anormal al ejercicio. Además, una publicación reciente postulaba que en pacientes sin criterios de riesgo de MS, la presencia de realce tardío>15% de la masa ventricular podría ser suficiente para indicar la implantación de un DAI12. Este trabajo ha recibido críticas debido a su diseño y a que sus conclusiones están basadas en los hallazgos de un número muy reducido de sujetos13.
Por el momento, no existen estudios que demuestren asociación entre el realce y la MS, aunque se ha comprobado que la presencia y extensión del realce sí se relaciona con el desarrollo de insuficiencia cardiaca.
En este escenario, y a falta de estudios multicéntricos bien diseña-dos, debe insistirse en que en la actualidad, el realce no debe emplearse en la estratificación de riesgo de MS.
Pruebas genéticas y cribado familiarEl manejo de la familia, el consejo genético y el cribado familiar son fundamentales en la atención de los pacientes con MCH.
Se recomienda realizar un análisis genético en los pacientes con MCH para posibilitar el cribado genético de sus familiares. Se le da una indicación I-
B, y se especifica que debe realizarse por «profesio-nales sanitarios entrenados» que formen parte de un «equipo multi-disciplinar», sin especificar quiénes deben integrar este equipo ni qué preparación se requiere para formarlos.
En nuestro país, esta recomendación debe apoyar la creación de consultas especializadas en cardiopatías familiares y la modificación del método de trabajo ante estas patologías. Hay que recalcar que es imprescindible el árbol familiar y el estudio familiar cuando se atiende pacientes con MCH. Esta «manera de trabajar» debería ser la habitual y no exclusiva de los centros de referencia.
Lo más relevante de este apartado es, sin duda, el mensaje claro y contundente sobre la utilidad de la genética. La realización de análisis genético es indicación clase I-B, pero teniendo en cuenta que, aunque permite asesoramiento genético, raramente ayudará al manejo clínico del probando. Los más beneficiados de la realización del estudio serán los familiares, ya que se podrán discriminar los portadores (que recibirán consejo genético y precisarán seguimiento) de los no porta-dores. También se resalta que el análisis genético es de dudosa utilidad en caso de probandos sin familiares accesibles o que no lo desean. En nuestro medio podemos destacar un estudio reciente que ha demostrado la utilidad de la realización de estudios genéticos en estos pacientes y sus familias, tanto desde un punto de vista clínico como económico14.
La guía aborda de forma prudente lo que constituye la práctica habitual hoy en día en cuanto al método de análisis genético. No se aboga por un método de análisis genético en particular aunque, en nuestra opinión, la Next Generation Sequencing parece la herramienta más adecuada en la actualidad, dado que permite estudiar de forma más eficiente un mayor número de genes. En la práctica clínica, la targeted Next Generation Sequencing, es decir, el estudio orientado hacia un grupo de genes de interés, más que el estudio del exoma completo, parece la estrategia más acorde con el conocimiento actual. El estudio del exoma puede implicar bajas coberturas (baja calidad del estudio) y el hallazgo de variantes en genes no relacionados con la MCH de difícil interpretación.
En consonancia con el documento de posicionamiento sobre estu-dios genéticos de la ESC15, la guía postula que, tras la identificación de una mutación causal en el probando, el análisis genético debe prece-der al estudio clínico de los familiares. Este es un aspecto, en nuestra opinión, erróneo. Se deben realizar ambos simultáneamente y nunca dejar de realizar el estudio clínico, puesto que en ocasiones el feno-tipo hallado en los familiares puede apoyar (o no) la patogenicidad de la mutación documentada en el probando. Además, alrededor del 5% de los pacientes con MCH portan 2 mutaciones, lo que puede tener trascendencia en el estudio familiar. Nuestra experiencia señala que obviar el estudio clínico sistemático puede resultar, en algunos casos, en errores diagnósticos.
Respecto al cribado clínico y genético en familiares menores de edad, el mensaje de la guía es poco concreto, con una indicación IIa y estableciendo la edad de 10 años como el punto a partir del cual se debe iniciar el seguimiento clínico y se puede plantear realizar el estu-dio genético. El espíritu de esta última recomendación se sustenta en la visión de que no es procedente realizar estudio genético en menores si un resultado positivo no modificará la actitud clínica y puede existir un impacto psicológico sobre el entorno familiar y el menor que puede condicionar su desarrollo. La opinión del grupo de trabajo en cuanto a este punto es divergente y no hay una evidencia clara que sustente cuál es la forma más adecuada de actuar en esta situación.
Sin embargo parece obvio que establecer la edad de 10 años como dintel de protección del menor no resuelve la controversia. El grupo de trabajo recomienda individualizar cada caso a la luz de las circuns-tancias particulares y el contexto familiar para establecer la idoneidad de la determinación y siempre tras llevar a cabo un asesoramiento genético adecuado.
Con respecto a la posibilidad de ofrecer diagnóstico genético pre-natal o preimplantacional, dado que la expresividad de la MCH es muy variable, se considera que el diagnóstico prenatal raramente estará indicado. Hay que reconocer que el marco legal de estas técnicas varía en cada país, de forma que es complicado establecer reco-mendaciones. Según el marco legal vigente en España para el diag-nóstico preimplantacional (detección de enfermedades hereditarias graves, de aparición precoz y sin tratamiento curativo) se requerirá habitualmente la aprobación previa de la Comisión Nacional de Reproducción Asistida.
Finalmente, y en relación con los estudios genéticos, cabe destacar 2 recomendaciones adicionales de la guía. La primera es la realización del estudio genético en pacientes fallecidos. En este caso, la indica-ción es solo IIa, lo cual llama la atención, pues el beneficio para los familiares es el mismo que en el caso de sujetos vivos (indicación grado I). En nuestra opinión, la indicación debe ser igualmente grado I. Además es importante que en nuestro país se creen protocolos conjuntos de actuación entre forenses-clínicos que permitan la conservación y posterior estudio de muestras de pacientes fallecidos súbitamente en los que la autopsia detecta una MCH. Existen ya protocolos al efecto en diversas autonomías y sería apropiado extender esta forma de trabajo a todo el territorio nacional.
Manejo de la obstrucciónEn este apartado, la guía básicamente consolida las recomendaciones vigentes aportando un abordaje sistemático para facilitar el manejo de la obstrucción. Además establece algunas aportaciones novedosas que es conveniente destacar.
Tratamiento médicoLos bloqueadores beta a dosis máximas toleradas constituyen el pilar del tratamiento de la obstrucción sintomática. Se hace mención especial al propanolol, pese a que no hay estudios comparativos que permitan recomendar un bloqueador beta en particular. Como novedad, la guía incluye el sotalol por su capacidad de tratar la obstrucción y las arritmias. La disopiramida queda establecida como el segundo escalón del tratamiento, siempre con bloqueador beta (no sotalol) o en ocasiones junto con verapamilo. Es importante monitorizar el intervalo QTc, que no debe superar 480ms, y está contraindicada con otros antiarrítmicos.
Habitualmente, en los pacientes con obstrucción, la fibrilación auricular (FA) se tolera mal, por lo que es importante restablecer el ritmo sinusal (IIa-C). La digoxina está contraindicada para el tratamiento de la FA en pacientes con obstrucción (III-C), aunque esta recomendación se basa en datos clínicos muy limitados.
Tratamiento invasivoSe subraya la importancia de la discusión de los casos en equipos multidisciplinares (heart team) y se hace hincapié en la experiencia de los profesionales que deben realizar un mínimo de 10 intervenciones anuales (20 por operador en la guía ACC/AHA3). Esto no es aplicable en absoluto a nuestro país, donde los estudios publicados indican muchísimos menos casos por centro y operador16. La única solución sería que los circuitos referenciadores fuesen de obligado cumplimiento, algo en lo que siempre se ha insistido y nunca se ha ejecutado en nuestro medio.
La indicación de tratamiento invasivo permanece invariable en pacientes con obstrucción significativa (gradiente>50mmHg basal o provocado) y síntomas limitantes (clase III-IV de la New York Heart Association) o sincope recurrente en relación con la obstrucción (I-B y IIa-C, respectivamente).
La ablación septal alcohólica se sitúa, por primera vez, como alternativa a nivel de la miectomía en centros expertos (I-B). No hay ensayos aleatorizados, pero los metanálisis han demostrado eficacia similar y complicaciones comparables. La ablación septal alcohólica tiene una tasa mayor de bloqueo auriculoventricular que la cirugía (el 12 frente al 5%).
La miectomía sería de elección en pacientes con hipertrofia grave (> 30mm), fibrosis extensa en la resonancia o en niños y adolescentes. En los casos de hipertrofia ligera (< 16mm) se recomienda valorar marcapasos o cirugía valvular mitral.
Existen algunos ensayos aleatorizados y controlados acerca del valor de la estimulación con marcapasos que demuestran reducción de la obstrucción con mejoría clínica variable. El beneficio de la estimulación bicameral parece ser mayor en sujetos mayores de 65 años; sin embargo, la guía no especifica qué grupos de pacientes podrían beneficiarse más de esta técnica (a diferencia de la guía ACCF/AHA4). Los autores dejan la indicación de marcapasos para pacientes con obstrucción sintomática que rechazan miectomía o ablación septal alcohólica, con indicación de marcapasos o que precisan un DAI (IIb-C).
Obstrucción medioventricularPor primera vez se dedica un apartado a la obstrucción mesocavi-taria, que aparece en el 10% de los pacientes, de los que una cuarta parte desarrollan aneurismas apicales. Los aneurismas apicales parecen asociarse a un mayor riesgo de arritmias ventriculares y de embo-lización. A pesar de lo anterior, no se recomienda indicación de DAI en ausencia de otros predictores de riesgo. Existe poca experiencia con la miectomía transaórtica o transapical para el tratamiento de la obs-trucción medioventricular.
MANEJO DE SÍNTOMAS EN AUSENCIA DE OBSTRUCCIÓNEn pacientes con una fracción de eyección del ventrículo izquierdo
<50% se asumen las recomendaciones de la guía de insuficiencia cardiaca respecto al tratamiento farmacológico17. Cabe destacar que la recomendación asume beneficios en términos de mortalidad, hospi-talizaciones y mejoría sintomática, sin evidencia probada en este grupo específico de pacientes. Se hace referencia a la complejidad en el manejo de estos pacientes, al no tolerar habitualmente dosis elevadas de vasodilatadores y diuréticos debido a la presencia de volúmenes ventriculares reducidos.
En relación con la terapia de resincronización se asumen las recomendaciones de las guías generales de estimulación (IIa-C). Además se establece una recomendación IIb para el caso de pacientes en clase funcional II-IV, fracción de eyección<50%, QRS>120ms y bloqueo de rama izquierda; esta controvertida recomendación se basa en los resultados de un pequeño estudio observacional unicéntrico18. En cualquier caso es destacable que se establezcan recomendaciones referentes a la terapia de resincronización, no presentes en la guía ACCF/AHA4.
La guía no menciona, y por tanto no se posiciona, nuevos fármacos antianginosos como la ivabradina y la ranolazina.
ARRITMIAS AURICULARESLa FA es una arritmia frecuente en los pacientes con MCH, con una prevalencia del 22,5% y una incidencia anual del 3,1% según un metanálisis recientemente publicado19. En este trabajo se identifica la edad y la dilatación AI como los predictores más importantes de FA19. El accidente cerebrovascular es una complicación frecuente. La prevalencia de accidentes cerebrovasculares en pacientes con MCH y FA en esta revisión fue del 27,1%, con una incidencia anual del 3,8%19.
No se incluyó a los pacientes con MCH en los estudios de estratifi-cación de riesgo embólico en la FA no valvular. El riesgo embólico en la MCH con FA es equivalente a un CHA2DS2-VASc (insuficiencia cardiaca congestiva, hipertensión, edad ≥ 75 [doble], diabetes, ictus [doble], enfermedad vascular y categoría de sexo [mujeres]) de 3, a pesar de ser una población mucho más joven. Los autores de la guía, no consideran aplicable el CHA2DS2-VASc en la MCH. En cambio, sí se aconseja la puntuación HAS-BLED (hipertensión, función renal/hepá-tica anormal, ictus, antecedentes de hemorragia o predisposición a ella, labilidad de la razón internacional normalizada, edad mayor 65 años y toma concomitante de fármacos o alcohol) para estimar la tasa de complicaciones hemorrágicas.
A partir de estos datos, la guía emite las primeras recomendacio-nes sobre FA en la MCH, de gran relevancia clínica:
- •
Es importante objetivar la presencia de FA de forma precoz, si es preciso por medio de Holter-electrocardiogramas periódicos (cada 6-12 meses) en pacientes con dilatación AI (> 45mm) (IIa-C).
- •
No se debe emplear la puntuación CHA2DS2-VASc y se debe ofrecer anticoagulación a todos los pacientes con MCH y FA en cualquiera de sus formas (I-B).
No hay alusiones al autocontrol de la anticoagulación y, en cambio, sí se recoge el uso de nuevos anticoagulantes como alternativa cuando hay dificultadescon los antagonistas de la vitamina K. El uso de ácido acetilsalicílico y clopidogrel puede considerarse cuando hay imposi-bilidad o rechazo del tratamiento anticoagulante, a pesar de que no existen estudios sobre su efectividad en MCH (IIa-B).
Cabe destacar que no se hace ninguna recomendación a la hora de plantear anticoagulación en pacientes con gran dilatación de aurícula izquierda, pese a que es una práctica extendida en diversos centros de referencia. La ausencia de datos sólidos que apoyen esta práctica clínica, probablemente ha motivado este proceder en la guía.
En cambio, sí se comenta la opción de la ablación de FA para casos seleccionados (AI no muy dilatada) refractarios a tratamiento antia-rrítmico (IIa-B), aunque no deja claro la actitud que se debe seguir en
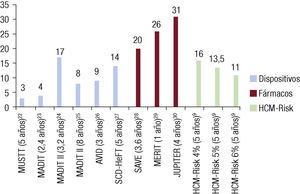
Esta aproximación supone un avance muy relevante en el campo y en el cálculo individual del riesgo de cada paciente y permite poner en perspectiva el número de pacientes a los que hay que implantar un DAI para evitar una MS22 (figura 1)23–31. La fórmula está disponible online22 y se ha creado una versión para dispositivos móviles.
.")
Dado que en menores de 16 años no se puede utilizar la fórmula HCM Risk-SCD (hypertrophic cardiomyopathy Risk-sudden cardiac death), se recomienda considerar la implantación de DAI cuando el número de factores de riesgo infantil es ≥ 2 (IIa-C) y valorarla en pre-sencia de 1 factor (IIb-C). Los factores de riesgo en edad pediátrica son: hipertrofia ≥ 30mm o z-score ≥ 6, taquicardia ventricular no sostenida, síncope inexplicado o MS en familiares. Debe tenerse en cuenta, y así lo señala la guía, que no existe apenas información relativa a pacientes con MCH menores de 8 años.
PREVENCIÓN DE LA MUERTE SÚBITAEste es uno de los capítulos más novedosos e importantes de la guía. Incluye información relevante de un estudio reciente donde se propone y se valida una fórmula para la estratificación del riesgo de MS9.
La guía europea previa consideraba que los pacientes que acumulaban 2 o más factores de riesgo clásicos eran de alto riesgo y, por tanto, se recomendaba implantar un DAI en prevención primaria2. La guía ACCF/AHA3 es menos exigente y considera de alto riesgo a pacientes con al menos un factor de riesgo. Se ha demostrado que la capacidad predictiva a 5 años de ambas guías es moderada, con un área bajo la curva de 0,64 y 0,63 para las guías ACC/ESC y ACCF/AHA, respectivamente20.
La nueva fórmula propuesta es resultado de un complejo análisis estadístico a partir de una población de 3.675 pacientes con MCH de 6 centros europeos, entre los que se incluyen 2 centros españoles9. Cada uno de los factores tiene un «peso» específico relativo. Se incluye la edad, el síncope inexplicado, el gradiente obstructivo, los antece-dentes de MS en familiares de primer grado, la presencia de taquicardia ventricular no sostenida, el diámetro de la AI y el máximo grosor ventricular. Alguno de los factores de riesgo clásicos, como la respuesta presora anormal, queda excluido y hay 2 nuevos: la edad y el diámetro de la AI. Hay otros posibles factores de riesgo que no se evaluaron, como la presencia de fibrosis en la resonancia, la isquemia miocárdica o las alteraciones genéticas.
Las recomendaciones para la implantación de DAI son prevención secundaria (I-B) o riesgo estimado de MS anual elevado (≥ 6%) (IIa-B). Debe evaluarse individualmente a los pacientes con un riesgo intermedio de entre el 4-6% anual (IIb-B). La fórmula no se ha comprobado y, por tanto, no debe emplearse en menores de 16 años, atletas, fenocopias (Fabry) y casos sindrómicos (p. ej., Noonan), y ofrece resultados paradójicos con hipertrofias extremas (> 35mm). Conviene destacar, además, que los individuos mayores de 65 años están apenas representados en el trabajo del que nace la fórmula y que debe usarse con precaución en pacientes sometidos a ablación septal alco-hólica/miectomía.
Aunque es conveniente que la fórmula se valide en múltiples poblaciones, ya se ha presentado el primer trabajo validatorio con excelentes resultados y con mejor capacidad discriminativa que las guías anteriores21.
REPRODUCCIÓN Y CONTRACEPCIÓNEs un apartado extenso y novedoso, aunque todas las recomenda-ciones tienen un nivel de evidencia C. Se propone el uso de anticon-ceptivos orales como método anticonceptivo preferido (salvo en la MCH con alto riesgo tromboembólico) y el dispositivo intrauterino como alternativa válida.
Se hace referencia expresa al riesgo de retención de fluidos y de tromboembolia venosa que puede suponer la fertilización in vitro, por lo que no debería realizarse en caso de insuficiencia cardiaca, FA o fisiología restrictiva.
En general, el embarazo puede desarrollarse con normalidad; res-pecto al parto, se optará por parto vaginal, salvo casos de riesgo por obstrucción grave, insuficiencia cardiaca o uso de anticoagulantes, en cuyo caso se prefiere una cesárea programada.
Es importante mantener una vigilancia estrecha de 24-48h tras el parto, por el riesgo de edema pulmonar debido a la redistribución de volumen.
SITUACIONES ESPECIALESEl último apartado de la guía recoge diversos escenarios en los que el diagnóstico y manejo de la MCH puede presentar particularidades. Aquí se recoge el diagnóstico diferencial con el corazón de atleta y la cardiopatía hipertensiva, el manejo de la hipertrofia septal basal del anciano y de valvulopatías asociadas.
En línea con la guía de valvulopatías32, no se recomienda la profi-laxis antibiótica de endocarditis sistemática en los pacientes con MCH. Aunque no hay datos sólidos que apoyen la profilaxis antibiótica es conveniente señalar que, al menos en los pacientes con obs-trucción, esta visión genera controversia en el campo.
Por último hay que destacar que esta guía no establece recomen-daciones claras en cuanto a la práctica deportiva recreacional (una consulta frecuente en estos pacientes) y que sigue sin haber acuerdo sobre la práctica deportiva competitiva en portadores sin expresión fenotípica de la enfermedad (IIb-C). En recomendaciones previas no estaba permitido por la ESC, pero sí por la AHA/ACC.
CONCLUSIONESLa guía de la ESC acerca del diagnóstico y manejo de la MCH es la más reciente actualización sobre el tema. En este documento encon-tramos información general y aspectos fundamentales del manejo de los pacientes con MCH, con una clasificación de las recomendaciones en 4 categorías que permite al cardiólogo clínico establecer la con-ducta más apropiada con cada paciente.
Los puntos más destacables de la guía hacen referencia al diagnós-tico etiológico de la MCH, los estudios genéticos, el manejo estructu-rado de la obstrucción del tracto de salida del ventrículo izquierdo, las arritmias auriculares y la prevención de la MS. Su contenido debe alentar la creación de unidades especializadas en la atención a estos pacientes y sus familias.
ANEXO. AUTORESGrupo de Trabajo de la SEC para la guía de la ESC 2014 sobre el diagnóstico y manejo de la miocardiopatía hipertrófica: Pablo Garcia-Pavia (Coordinador), Josep Comín-Colet (Coordinador), Roberto Barriales-Villa, Vicente Climent, Enrique Galve, José Manuel García-Pinilla, Juan Ramón Gimeno-Blanes, Tomás Ripoll-Vera y Maite Tomé.
Revisores expertos para la guía de la ESC 2014 sobre el diagnóstico y manejo de la miocardiopatía hipertrófica: Luis Almenar, Luis Alonso-Pulpón, Manuel Anguita, Begoña Benito Villabriga, Marta Cobo-Marcos, Juan Delgado, Gonzalo Guzzo-Merello, Jose Luis Lambert, José López-Haldón, José Julián Rodríguez Reguero, Sonia Ruiz, Joel Salazar-Mendiguchía, Helena Tizón y Esther Zorio.
Comité de Guías de la SEC: Manuel Anguita Sánchez (Presidente), Ángel Cequier-Fillat, Lina Badimón Maestro, José Antonio Barrabés Riu, Josep Comín-Colet, Ignacio Fernández Lozano, José Juan Gómez de Diego, Manuel Pan Álvarez-Osorio, Luis Rodríguez Padial, José Alberto San Román Calvar y Pedro Luis Sánchez Fernández.
CONFLICTO DE INTERESESNinguno.
REFERENCIA NO CITADA[22].