
Presentamos una revisión de algunos de los artículos más significativos publicados en el área de las cardiopatías congénitas y la cardiología pediátrica durante 2010 y hasta septiembre de 2011, con especial interés en los relacionados con los cambios demográficos que se han producido en esta población y con la necesidad de realizar la transición de estos pacientes desde los servicios de cardiología pediátrica a los de adulto. Ello ha dado lugar a la aparición de nuevas áreas de interés, como el embarazo en mujeres con una cardiopatía congénita y el papel que los factores genéticos pueden tener en la etiología y la transmisión de determinadas anomalías. Asimismo, y con el objetivo de precocidad diagnóstica y, de ser posible, terapéutica, se revisan algunos artículos relacionados con la cardiología fetal. Seguidamente se mencionan las nuevas aportaciones en el síndrome de Eisenmenger y las arritmias, así como en técnicas de imagen, cateterismo intervencionista y trasplante cardiaco; finalmente se alude a la nueva versión de las guías de práctica clínica sobre el manejo del paciente adulto con una cardiopatía congénita y a la recientemente publicada guía sobre el embarazo de mujeres con cardiopatía, ambas procedentes de la Sociedad Europea de Cardiología.
Palabras clave
Abreviaturas
AV: auriculoventricular.
BAVC: bloqueo AV completo.
CC: cardiopatía congénita.
CF: clase funcional de la New York Heart Association.
DAI: desfibrilador automático implantable.
VD: ventrículo derecho.
INTRODUCCIÓN
Presentamos una revisión de algunos de los artículos más importantes sobre cardiopatías congénitas (CC) y cardiología pediátrica publicados en 2010 y hasta septiembre de 2011. De particular interés son los artículos que tratan sobre los cambios demográficos producidos en esta población, así como sobre la necesidad de hacer el proceso de transición de estos pacientes de los servicios de cardiología pediátrica a los de adultos, con la aparición de nuevas áreas de interés, como el embarazo de mujeres con CC o el papel que los factores genéticos pueden tener en la etiología y la transmisión de ciertas anomalías. Además, con el objetivo de un diagnóstico y, de ser posible, un tratamiento precoces, se revisan algunos artículos relacionados con la cardiología fetal. A continuación se describen algunos nuevos aportes científicos para el síndrome de Eisenmenger (SE) y las arritmias, así como en técnicas de imagen, el cateterismo intervencionista y el trasplante cardiaco. Finalmente, se revisa la nueva versión de las guías clínicas sobre el tratamiento de pacientes adultos con cardiopatías congénitas y las guías recientemente publicadas sobre el embarazo de mujeres con cardiopatía, ambas de la Sociedad Europea de Cardiología.
ESTUDIOS DEMOGRÁFICOS
La incidencia de CC al nacimiento sigue siendo materia de cuestión, pero su conocimiento es esencial para la planificación de los recursos sanitarios. Recientemente se han publicado los datos de la European Surveillance of Congenital Anomalies, un registro poblacional realizado en 16 países de Europa que cubre una población de 3,3 millones de nacimientos1. La incidencia total de CC fue del 0,8%, de los cuales el 3,6% fueron muertes perinatales y el 5,6%, interrupciones del embarazo por anomalía fetal grave. En conjunto, el 20% de las CC y el 40% de las CC graves se diagnosticaron prenatalmente.
Los avances en cardiología y cirugía cardiaca pediátrica han condicionado un cambio en la prevalencia de las CC en la edad adulta. Moons et al2 han analizado la proporción de pacientes, nacidos entre 1990 y 1992 en Bélgica, que sobrevivieron hasta los 18 años de edad en comparación con los pacientes nacidos en las décadas previas, en función del tipo de CC. La supervivencia actual hasta los 18 años fue del 89%, comparado con el 81% de los individuos nacidos entre 1970 y 1974, el 82% de los nacidos entre 1975 y 1984 y el 87% de los nacidos entre 1985 y 1989 (p < 0,001). La supervivencia total fue > 96% en CC simples, pero < 90% en la coartación aórtica (89%), la tetralogía de Fallot (T4F) (78%), la transposición (71%), el ventrículo único (49%) y el síndrome del corazón izquierdo hipoplásico (SCIH) (7,5%). No hubo diferencia significativas entre periodos en pacientes con CC simples, pero hubo un aumento progresivo de la supervivencia en CC moderadas (p < 0,0006) y complejas (p < 0,0002).
Un dato notable es que la tasa de mortalidad en CC está disminuyendo progresivamente. Entre 1979 y 1997, la mortalidad ajustada por edad relacionada con CC había disminuido en Estados Unidos en un 39%. Esta tendencia se ha mantenido en los últimos años y entre 1999 y 2006 la tasa de mortalidad se ha reducido en otro 24%3. Partiendo de una tasa de mortalidad de 2,5/100.000 habitantes en 1979, se redujo a 1,9/100.000 en 1997 y a 1,2/100.000 en 2006. La disminución de la mortalidad es mucho más notable en cardiopatías complejas como T4F, transposición y canal auriculoventricular (AV) completo.
Lo que ha ocurrido es una desviación de la mortalidad desde el niño hacia el adulto4. La proporción de muertes en la infancia disminuyó marcadamente entre 1987 y 2005 en la población de Quebec, con una reducción en mortalidad que excedía la de la población general. La distribución de la edad de muerte cambió de un patrón bimodal (con un alto pico inicial en el primer año de vida y un segundo pico más bajo durante la edad adulta) a un patrón unimodal siguiendo una distribución similar a la de la población general. De nuevo, estos cambios son mucho más evidentes en CC complejas. En estos pacientes, la mediana de la edad de muerte aumentó desde los 2 años entre 1987 y 1993 a los 23 años entre 1999 y 2005 (p < 0,001).
Estos estudios están basados en análisis poblacionales que utilizan bases de datos administrativas, pero cuyos registros de mortalidad son imprecisos5; se necesitan registros clínicos que recojan de forma más adecuada la mortalidad y el modo de muerte de los pacientes con CC. El registro holandés CONCOR6 comenzó en 2001, y hasta 2010 se ha incluido aproximadamente a 12.000 pacientes con CC mayores de 18 años. Durante este periodo, la mortalidad total fue de 8/1.000 pacientes-año. Obviamente, la edad fue un predictor de mortalidad, pero había un exceso de mortalidad respecto a la población general, en todos los grupos de edad y particularmente entre los más jóvenes. La causa de muerte fue cardiovascular en el 77% de los casos, con la insuficiencia cardiaca (26%) y la muerte súbita (19%) como principales modos de muerte. Los principales predictores de mortalidad fueron la gravedad de la CC, el número de intervenciones y el número de complicaciones, como endocarditis, arritmias auriculares o ventriculares, trastornos de la conducción, infarto de miocardio o hipertensión pulmonar (HP).
En el último año se han publicado dos importantes marcadores de mortalidad en la población adulta con CC. En un estudio7, la hiponatremia, definida como una concentración de sodio < 136 mmol/l, identificó a pacientes con triple riesgo de muerte; esta relación fue independiente de la edad, cirugía previa, clase funcional (CF) de la New York Heart Association (NYHA), función del ventrículo sistémico, concentración sérica de creatinina y uso de diuréticos (hazard ratio [HR] ajustada = 2,82; intervalo de confianza del 95% [IC95%], 1,72-4,63; p < 0,0001). En otro estudio del mismo grupo8, se analizó la relación entre los péptidos natriuréticos cerebral y auricular y el riesgo de muerte a medio plazo. Ambos péptidos fueron fuertes marcadores de mortalidad (respectivamente, por cada 100 pg/ml: HR = 1,80; IC95%, 1,38-2,34; p < 0,0001, y HR = 1,21; IC95%, 1,12-1,32; p < 0,0001).
Transición del niño al adulto
Los estudios demográficos muestran que el número de adultos con CC es ya mayor que el de niños; este aumento es especialmente significativo en CC complejas. Esta avalancha plantea necesidades asistenciales no consideradas hasta ahora9. Persiste la controversia sobre si se debe planear estas necesidades asistenciales en un medio pediátrico, para adultos o compartido10, pero hay amplio acuerdo sobre la necesidad de realizar programas de transición desde el ambiente pediátrico al adulto. Recientemente, la American Heart Association ha elaborado unas guías clínicas de cómo realizar la transición en adolescentes con CC. En este interesante documento, se abordan todas las recomendaciones relacionadas con este tópico11. El inicio del proceso depende de las características médicas y del desarrollo de cada individuo, pero se recomienda que comience a partir de los 12 años. Durante la niñez, el paciente debe ir involucrándose progresivamente en discusiones abiertas sobre su diagnóstico, sus medicaciones y sus limitaciones físicas. En la adolescencia, se debe tratar los temas de conducta cardiosaludable y los riesgos del tabaco, alcohol y otras drogas. Los consejos vocacionales y ocupacionales, así como de control de la natalidad, embarazo, genética y pronóstico a largo plazo, deben prolongarse hasta la vida adulta. La tabla 1 resume los tópicos principales que deben conocer los adolescentes y sus familiares antes de abandonar el ambiente pediátrico.

Sin embargo, en niños con CC, la transición a un medio adulto es especialmente complicada por las dificultades de obtener un cuidado especializado de igual calidad que el que obtenían en el ambiente pediátrico. Estudios realizados en Canadá, Estados Unidos, Alemania y Reino Unido han señalado como uno de los problemas principales de la transición el abandono del seguimiento especializado cuando se termina la dependencia del ambiente pediátrico. Estos datos contrastan con los recientemente publicados por el grupo SWITCH de la Universidad de Leuven (Bélgica)12. Entre 794 pacientes con CC mayores de 21 años, esos autores registran falta de seguimiento de sólo el 7%, incluido un 2,7% de casos con CC complejas. Las razones que explicarían este nivel de excelencia pueden ser: a) centralización del seguimiento de las CC; b) localización en el mismo edificio de los programas de adultos y pediátricos; c) compartir la historia clínica, el sistema de información y las bases de datos; d) vigilancia médica que incluye cartas recordatorias de las visitas médicas programadas; e) cobertura sanitaria de toda la población; f) acceso directo de los pacientes al hospital terciario (sin necesidad de orden de asistencia), y g) país pequeño con alta densidad de población y buenas comunicaciones. Excepto esta última condición, las demás características pueden ser similares en nuestro país y deberíamos aprender de estos vecinos comunitarios la manera de conseguir este nivel de excelencia.
Embarazo
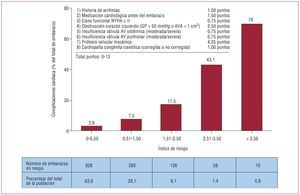
Dados los cambios epidemiológicos, cada vez más mujeres con una CC alcanzan la edad de procrear, lo que introduce la necesidad de valorar y aconsejar sobre el riesgo que pueden comportar el embarazo y el parto. En este contexto, creemos importante resaltar el estudio de Drenthen et al13, dirigido específicamente a la búsqueda de factores predictores de complicaciones ante el embarazo de una mujer con CC. En 2001, Siu et al14 publicaron un estudio prospectivo y multicéntrico (CARPREG) sobre mujeres con enfermedad cardiaca, no necesariamente congénita, e identificaron una serie de variables previas al embarazo a través de las cuales establecieron una puntuación para calcular el riesgo de complicaciones cardiacas maternas durante o inmediatamente después de una gestación. En el estudio de Drenthen13, aunque retrospectivo, se observó que el índice CARPREG sobrevaloraba el riesgo de complicaciones en esta población y elaboraron un nuevo índice de riesgo (IR) (fig. 1) que, si bien precisa de futuras validaciones, no deja de proporcionar información que hay que tener en cuenta. En concordancia con el estudio CARPREG, fueron predictores independientes de complicaciones maternas una CF > II, la presencia de lesiones obstructivas del tracto de salida ventricular izquierdo (TSVI) y antecedentes de arritmias, pero también lo fueron una insuficiencia moderada-grave de la válvula AV sistémica o pulmonar, la presencia de una prótesis mecánica valvular y la existencia de una CC cianótica al nacer (aunque corregida). En cuanto al riesgo en el hijo, factores como el tratamiento farmacológico cardiaco, la existencia de una CC cianótica al nacer y la presencia de una prótesis mecánica valvular se detectaron como nuevos factores de riesgo de complicaciones fetales o neonatales. En cambio, y a diferencia de lo que señalan Siu et al14, en el estudio de Drenthen13 la CF > II y la presencia de cianosis u obstrucción del TSVI no se asociaron con este tipo de complicaciones. En cualquier caso, si bien disponemos de un nuevo IR aplicable a mujeres con una CC, creemos que es importante hacer énfasis en algunas de las limitaciones del estudio: retrospectivo, limitado a mujeres controladas en un centro terciario, ausencia de grupo control, etc., pero especialmente el hecho de que los factores de riesgo detectados dependen de la muestra estudiada; por ejemplo, no haber incluido a mujeres con HP lógicamente comporta la ausencia de dicha variable entre los factores de riesgo observados. Por eso se debe ser cauto antes de dar información en este contexto y no apoyarse únicamente en los IR publicados.

Figura 1. Índice de riesgo modificado para complicaciones cardiacas durante una gestación > 20 semanas, en mujeres con una cardiopatía congénita (expresado como porcentaje del total de embarazos de más de 20 semanas). AV: auriculoventricular; AVA: área valvular aórtica; GP: gradiente pico. Obtenido de Drenthen et al13.
Un tema menos evaluado es la posibilidad de que un embarazo, con la correspondiente sobrecarga hemodinámica mantenida durante unos meses, pueda comportar complicaciones a largo plazo, especialmente cambios estructurales o de la función ventricular. Fue el objetivo del estudio de Balint et al15 en el que, tras evaluar 405 embarazos en 318 mujeres con una CC y un seguimiento de 2,6 años, se detectó un 12% de complicaciones a largo plazo (> 6 meses posparto). Las más frecuentes fueron las arritmias, seguidas de insuficiencia cardiaca, pero cabe citar que se produjeron 3 muertes, una de ellas a los 2 años de una gestación sin problemas en una paciente con una T4F corregida, con insuficiencia pulmonar (IP) grave y disfunción de ventrículo derecho (VD). Un dato a resaltar es que, aunque la disfunción del ventrículo subpulmonar o la presencia de IP grave no se asociaron a un mayor número de eventos durante la gestación, a largo plazo sí se asociaron. Asimismo, tanto el índice de CARPREG como la presencia de complicaciones durante el embarazo mostraron ser predictores de eventos a largo plazo. En el análisis multivariable, la CF > II o la cianosis, la disfunción del ventrículo sistémico, la disfunción del ventrículo subpulmonar o la IP, la estenosis del TSVI y las complicaciones cardiacas antes o durante la gestación se asociaron a complicaciones tardías. A partir de dichos factores, se diseñó un nuevo IR para complicaciones tardías, cuyo carácter discriminatorio puede observarse en la figura 2.

Figura 2. Incidencia de eventos cardiacos tardíos después de una gestación en relación con un índice de riesgo materno. Los eventos cardiacos adversos se definieron como muerte cardiaca, parada cardiaca, edema pulmonar, taquiarritmia sostenida sintomática o bradiarritmia que requiera tratamiento o accidente cerebrovascular o accidente isquémico transitorio. Obtenido de Balint et al15.
GENÉTICA
En los últimos años ha habido grandes avances en el campo de la genética molecular y la biología del desarrollo pero, a pesar de estos progresos en la comprensión del desarrollo cardiaco y de la identificación de muchos genes relacionados con este, la etiología fundamental de la mayoría de los casos de CC permanece desconocida. Huang et al16 han revisado los mecanismos de la morfogénesis cardiaca, subrayan los descubrimientos más recientes sobre las causas genéticas de CC y apuntan posibles estrategias presentes y futuras para explorar dichas causas.
Andelfinger et al17 realizan una puesta al día de la evidencia actual en los conocimientos sobre los factores genéticos implicados en las CC, analizan la interacción potencial de tales factores en diferentes modelos y destacan algunas cuestiones que la investigación en genética nos puede aportar en relación con la investigación clínica y traslacional en los próximos años. En una exhaustiva puesta al día, Wessels et al18 analizan los genes de enfermedades monogénicas con mutaciones de alta penetrancia y mutaciones somáticas implicadas en CC que no forman parte de un síndrome.
La evolución del conocimiento en genética cardiovascular en los últimos 15 años ha refinado nuestro mecanismo de comprensión de los síndromes cardiacos hereditarios asociados con la muerte súbita cardiaca, y también ha modificado el enfoque clínico de estos pacientes y sus familias. Un amplio panel de expertos lo ha revisado recientemente y ha plasmado en un documento de consenso las recomendaciones sobre el papel de los distintos tests genéticos en diferentes contextos clínicos asociados con muerte súbita cardiaca19.
Cardiología fetal
Debido a la morbimortalidad de la transferencia transplacentaria de los anticuerpos maternos SS-A/Ro y SS-B/La, se han diseñado protocolos para el manejo de estos fetos de riesgo. El Doppler pulsado que mide el intervalo mecánico AV ha sido propuesto como método de vigilancia y detección precoz en los fetos con riesgo de bloqueo AV completo (BAVC). Sin embargo, en la actualidad no existen unas directrices estándar para los fetos expuestos a anticuerpos maternos, ni tampoco hay consenso sobre si se debe tratar o no la prolongación de la conducción AV. En un estudio prospectivo que incluyó a 165 fetos de 142 madres con anticuerpos anti-Ro/SSA y anti-La/SSB, Jaeggi et al20 estudian la prevalencia de la prolongación de la conducción AV utilizando Doppler pulsado en vena cava superior-aorta, mitral-TSVI y Doppler tisular de VD. De 150 fetos con conducción AV persistentemente normal durante un seguimiento semanal entre las semanas 19 (17-23) y 24 (23-35), un feto presentó BAVC en la semana 28 de gestación. De 15 fetos no tratados que presentaron BAV de primer grado o segundo grado Mobitz I, ninguno tuvo progresión del grado de bloqueo. De estos 15 fetos, 3 presentaron BAV de primer grado de neonatos, que se resolvió espontáneamente (2) o no progresó (1). Proponen que el tratamiento con dexametasona debe restringirse a fetos con BAV progresivo o con otros hallazgos, como fibroelastosis endocárdica o derrame pericárdico. En cambio, en prolongaciones aisladas del intervalo AV (hasta z-score 6) recomiendan una estrecha vigilancia de la progresión. Hacen hincapié en la variabilidad del observador al medir la conducción AV por los diversos métodos y en los pocos milisegundos que representa una desviación estándar (z-score). Concluyen que la prolongación del intervalo AV fetal no es predictivo de BAVC.
Se desconoce por qué sólo algunos de los fetos expuestos a autoanticuerpos sufren BAVC. En los embarazos de madres con anticuerpos anti-Ro y anti-La, la prevalencia de BAVC fetal es de un 1-5%, que aumenta a un 6-25% para las que tengan un niño previamente afecto. Jaeggi et al21, en un trabajo prospectivo con 186 fetos expuestos a anticuerpos, determinaron los niveles maternos de anticuerpos anti-Ro y anti-La. Todas las complicaciones cardiacas estaban asociadas a aumentos moderados (> 50 U/ml; 15%) o altos (> 100 U/ml; 85%) de anticuerpos maternos anti-Ro, independientemente de la titulación de anti-La. Sus hallazgos confirman que la cantidad de anticuerpos maternos, en lugar de sólo su presencia, se asocia con lesiones fetales. Señalan que para valorar el riesgo de BAVC es importante no sólo la presencia, sino también su concentración.
Nunca se ha logrado revertir un BAVC. Dos estudios multicéntricos evalúan el tratamiento preventivo de BAVC con inmunoglobulinas. Friedman et al22 incluyeron a madres con anticuerpos SSA/Ro que habían tenido previamente un hijo con cualquier tipo de bloqueo. Administraron inmunoglobulinas intravenosas (400 mg/kg cada 3 semanas) desde la semana 12 a la 24. De 20 gestantes, 3 fetos (en semanas 19, 20 y 25) sufrieron BAVC (15%), con controles previos del intervalo AV normales. En el estudio de Pisoni et al23 se incluyeron fetos de mujeres con anticuerpos anti-Ro y anti-La que habían tenido previamente un feto con BAVC. Utilizaron inmunoglobulinas a las mismas dosis y con los mismos intervalos, con un grupo control. Tres fetos del grupo tratado (20%) y 1 en el grupo control (11%) sufrieron BAVC. Ambos estudios concluyen que la administración de inmunoglobulinas a esas dosis no evita un nuevo feto con BAV.
La valvuloplastia aórtica fetal es técnicamente posible y en un tercio de los fetos afectados se han encontrado signos suficientes de mejoría hemodinámica del corazón izquierdo para alterar la historia natural y lograr una circulación biventricular. Arzt et al24 han descrito su experiencia con la valvuloplastia aórtica en fetos con estenosis aórtica valvular crítica. El 66,7% de estos procedimientos (16 de 24) tuvo éxito. En 8 de 24 (33%), el procedimiento falló (no se consiguió atravesar la válvula). Se produjeron 3 muertes intrauterinas. En el grupo de 16 fetos con éxito técnico, hubo 1 muerte intrauterina y 15 nacidos vivos, de los que 10 tuvieron circulación biventricular y 5, univentricular. Los autores concluyen que la valvuloplastia aórtica fetal podría llevarse a cabo en fetos seleccionados con estenosis aórtica crítica que evolucionan a SCIH, con un resultado biventricular en dos tercios de los pacientes.
Síndrome de Eisenmenger
La HP es una de las complicaciones más graves de los pacientes con CC con cortocircuito, con la posible evolución a SE: progresiva elevación de las resistencias vasculares pulmonares e inversión del cortocircuito, con aparición de hipoxemia arterial y cianosis. Únicamente disponemos de un estudio aleatorizado y controlado con placebo (BREATHE 5), que demuestra el efecto beneficioso del bosentán en este tipo de pacientes, pero en el último año se han publicado diversos estudios que, si bien menos ambiciosos, ponen de manifiesto el efecto que otros fármacos pueden tener en enfermos con una CC e HP grave. Si nos ceñimos a los antagonistas de la endotelina, cabe citar el estudio de Zuckerman et al25, en el que, tras la administración de ambrisentán a una cohorte de pacientes consecutivos con SE, se observó una mejoría de la capacidad funcional a corto plazo (seguimiento medio, 163 ± 57 días) y su estabilización a largo plazo (seguimiento medio, 2,5 ± 0,5 años), a la vez que el mantenimiento de la saturación arterial de O2 en reposo y durante el ejercicio, la CF y la hemoglobina, tanto a corto como a largo plazo. Dato importante es que no se observó elevación de las enzimas hepáticas en ningún paciente y que, aunque se produjeron 3 muertes, sólo en 1 caso se produjo estando el paciente en tratamiento con ambrisentán.
En cuanto a los inhibidores de la fosfodiesterasa 5, Tay et al26 han comunicado mejoría de la CF, la capacidad de ejercicio y la calidad de vida (cuestionario CAMPHOR) después de tratar con sildenafilo durante 3 meses a 12 pacientes con SE en CF III. A ello hay que añadir que no hubo disminución de la saturación arterial de O2 ni efectos adversos importantes. Estos hallazgos estarían en consonancia con los publicados por D'Alto et al27, que demuestran el efecto beneficioso de la adición de sildenafilo al bosentán en pacientes con dicha enfermedad. En ese estudio, a 32 pacientes con HP asociada a una CC (28 con SE) y tratados con bosentán, tras observarse un empeoramiento clínico y practicarse un estudio hemodinámico, se añadió sildenafilo durante 6 meses y se observó una mejoría de la CF, la saturación arterial de O2, la capacidad funcional, el índice de Borg, la concentración de la fracción aminoterminal del propéptido natriurético B (NT-proBNP) y de algunos paramétros hemodinámicos.
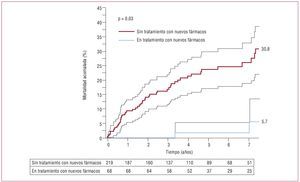
De especial interés es el estudio de Dimopoulos et al28, en el que —aparte del efecto beneficioso en cuanto a capacidad funcional, calidad de vida, parámetros hemódinamicos, etc., que estos nuevos fármacos producen en los pacientes con SE— se registró una mejora de la supervivencia. En dicho estudio se incluyó a 229 pacientes en CF ≥ III, la mayoría con una anatomía compleja (44,5%). Se contabilizaron 52 muertes durante un seguimiento medio de 4 años, pero sólo 2 en pacientes en tratamiento con alguno de estos nuevos fármacos. Ciertamente, como pone de manifiesto la figura 3, los pacientes en tratamiento mostraron un riesgo de muerte por cualquier causa significativamente menor, aunque pertenecían al grupo de mayor edad, más sintomáticos y con más posibilidades de recibir tratamiento anticoagulante y sufrir cuadros sincopales (HR bruta = 0,21; IC95%, 0,05-0,86; p = 0,03).

Figura 3. Curvas de supervivencia no ajustada (intervalo de confianza del 95%) según tratamiento con los nuevos fármacos (n = 287). Obtenido de Dimopoulos et al28.
ARRITMIAS
Las mejoras en el diagnóstico y la tecnología han conducido a un incremento en la implantación de DAI en niños y pacientes jóvenes. No obstante, las indicaciones en estos pacientes, especialmente en prevención primaria, siguen estando mal definidas.
Los datos sobre implantación de DAI en Estados Unidos29 en menores de 18 años muestran un aumento de 3 veces desde 1997 a 2006 (de 130 en 1997 a 396 en 2006). Los implantes con un diagnóstico concomitante de arritmia potencialmente mortal se redujeron del 77 al 45%, como reflejo del incremento de implantes en prevención primaria. El porcentaje de pacientes menores de 5 años tendió a aumentar (hasta un 10%).
En un estudio multicéntrico30 que incluyó a 210 pacientes menores de 30 años a los que se implantó un DAI —mediana de edad al implante, 15,4 (intervalo, 0,2-29,2) años—, la cardiopatía se clasificó como eléctrica (n = 92; 42%), miocardiopatía (n = 62; 30%) y CC (n = 58; 28%). Hay más descargas apropiadas en prevención secundaria (52%) que en prevención primaria (14%). En ambos grupos, aproximadamente el 25% de los pacientes recibieron descargas inapropiadas en los 5 años tras el implante. No hay ninguna diferencia de riesgo de recibir descargas inapropiadas entre las indicaciones de prevención primaria o secundaria. Los pacientes con CC son los más afectados por descargas inapropiadas. Los datos de ese trabajo demuestran un aumento de implantación en prevención primaria de un 15% antes de 1999 a casi el 50% actualmente. Este aumento puede ser reflejo del impacto de las pruebas genéticas en la toma de decisiones. También la tecnología de los dispositivos ha mejorado y hay un mayor reconocimiento del riesgo de muerte súbita en la población con CC de más edad. Definen prevención primaria como la ausencia de arritmia ventricular previa, muerte súbita abortada o síncope no explicado en paciente considerado de alto riesgo.
En el artículo de Horner et al31, 51 pacientes (niños y sobre todo adultos jóvenes) de 451 con síndrome de QT largo (SQTL) comprobado genéticamente (14 SQTL1, 22 SQTL2 y 15 SQTL3) recibieron un DAI entre 2000 y 2010. Es importante destacar que no hubo muertes relacionadas con SQTL entre los 408 pacientes tratados sin DAI. Se puede tratar eficazmente sin DAI a la gran mayoría de los pacientes con SQTL. Aunque la frecuencia de implante de DAI es mayor entre los pacientes con QTL3, la mayor tasa de descargas «salvavidas» se produjo en mujeres con QTL2.
Imagen cardiaca
La ecocardiografía es, sin duda, la técnica de imagen que más información aporta sobre estos pacientes, pero cada vez con mayor frecuencia es necesario recurrir a otros métodos diagnósticos, como la resonancia magnética (RM) o la tomografía computarizada (TC). Existen muchos trabajos que muestran la utilidad de estas técnicas radiológicas en la valoración de los pacientes con CC, pero se echaba de menos la sistematización de sus indicaciones. Broberg et al32 han publicado recientemente una excelente revisión sobre los avances de la ecocardiografía, RM y TC multidetectores durante la pasada década, resaltando la utilidad de estas herramientas en el manejo clínico de los pacientes con CC.
En 2010 se publicaron las recomendaciones de la ESC para la práctica de RM en CC del adulto, avalada por los grupos de trabajo respectivos33. Ese documento de consenso analiza las fortalezas y debilidades de esta técnica y provee información específica sobre su utilidad clínica en las CC. Sus principales ventajas son la definición de las conexiones anatómicas, la cuantificación de la función de ambos ventrículos, la medida de los flujos, la angiografía no invasiva y la valoración de la viabilidad miocárdica sin exposición a radiaciones ionizantes. La mayor limitación es que requiere formación específica, tanto para la adquisición como para la interpretación de las imágenes, y un conocimiento fisiopatológico profundo de las CC complejas y las modificaciones impuestas por los procedimientos terapéuticos, por lo que exige un largo periodo de formación y mucha experiencia. El documento proporciona una extensa tabla de protocolos de adquisición de imagen en las diferentes categorías diagnósticas como guía para el uso apropiado de esta técnica. La tabla 2 muestra las principales indicaciones de la RM recomendadas para adultos con CC.

Una de las principales indicaciones de la RM en CC es la valoración de la función del VD, sea este de localización subpulmonar o subaórtica o único. Alonso-González et al34 han publicado recientemente en REVISTA ESPAÑOLA DE CARDIOLOGÍA una excelente revisión sobre la anatomía del VD y los efectos adversos de su disfunción en pacientes adultos con CC. Numerosos estudios tratan de alcanzar el nivel de agudeza proporcionado por la RM utilizando las nuevas herramientas ecocardiográficas: Doppler tisular, stress-strain y, sobre todo, eco-3D. Van der Zwaan et al35 han comparado los volúmenes y la fracción de eyección (FE) del VD en 100 pacientes con diversas CC utilizando RM y eco-3D en tiempo real. Los valores de corte para el diagnóstico de disfunción del VD por eco-3D fueron 105 ml/m2 de volumen telediastólico, 54 ml/m2 de volumen telesistólico y 43% de FE. Utilizando estos valores de corte, la sensibilidad del eco-3D para determinar disfunción del VD fue del 95%, con una especificidad del 89% y un valor predictivo negativo del 99%.
CATETERISMO INTERVENCIONISTA
Aunque desde 1980 la implantación de stents para el tratamiento de la coartación aórtica se ha establecido como una alternativa a la cirugía, los resultados disponibles están condicionados por un corto seguimiento de los pacientes, así como una inadecuada aplicación de las técnicas de imagen para determinar el resultado anatómico y hemodinámico. Holzer et al36 firman el estudio multicéntrico más significativo publicado hasta la fecha, que demuestra buen resultado en el seguimiento a largo plazo (hasta 60 meses), con una incidencia de disección y aneurismas < 1%. Sin embargo, a pesar de ser una técnica eficaz, siempre ha existido la duda sobre los efectos hemodinámicos que pudiera tener la interposición de una estructura rígida como el stent en el arco aórtico y cómo puede afectar esa pérdida de distensibilidad de la pared vascular. Coogan et al37, mediante un modelo virtual computarizado usando RM y reconstrucción 3D, han demostrado que no existen alteraciones hemodinámicas significativas al implantar una estructura rígida en el arco aórtico.
La dilatación percutánea con balón de las estenosis subaórticas «tipo membrana» ha sido un tema muy controvertido en los últimos años. Sin embargo, Suárez de Lezo et al38 han descrito sus resultados a largo plazo con esta técnica, en la que han sido pioneros. Su serie consta de 76 pacientes (3-67 años) en los que se dilató con balón una estenosis subaórtica aislada. La reducción de gradiente pico inmediata fue de 70 ± 27 a 18 ± 12 mmHg (p < 0,001). No se objetivó ningún grado de insuficiencia aórtica significativo después del procedimiento. El periodo de seguimiento fue de 16 ± 6 años, 11 pacientes (15%) sufrieron reestenosis; 3 (4%), una estenosis «muscular», y en 1 paciente se produjo reestenosis por una nueva estructura membranosa. El 16% (12 pacientes) fueron redilatados en un plazo medio de 5 ± 3 años después del primer procedimiento, 4 (5%) fueron intervenidos quirúrgicamente durante el seguimiento; 58 pacientes (77%) están vivos y sin reintervención percutánea ni quirúrgica. El tamaño del anillo membranoso y su grosor no fueron factores anatómicos que condicionaran el éxito de la técnica a largo plazo.
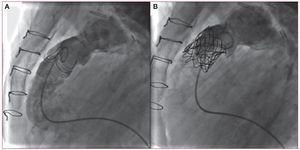
La implantación percutánea de una válvula pulmonar (IPVP) se ha consolidado como una opción eficaz para el tratamiento de la disfunción del tracto de salida ventricular derecho, ya se trate de conductos valvulados, prótesis biológicas (fig. 4) o, en algunos casos favorables, ampliaciones quirúrgicas con parche. Sin embargo, los efectos de la IPVP sobre el VD todavía no se han establecido definitivamente. Lurz et al39 han presentado una serie de 65 pacientes en los que se realizó una IPVP con prótesis tipo Melody (Medtronic). Se dividió a los pacientes en dos grupos: con estenosis pulmonar (n = 35) y con insuficiencia como lesión predominante (n = 30). Se estudió a ambos grupos mediante ergometría y RM en situación basal, y al mes y al año después del procedimiento. En el primer mes, en ambos grupos hubo una disminución significativa del volumen telediastólico del VD, con un aumento de la FE del VD sólo en el grupo de estenosis (el 51% ± 11% frente al 58% ± 11%). Después de 1 año, no hubo ningún cambio significativo en el estudio de RM en ningún grupo respecto al volumen o la función del VD. En cuanto a la prueba ergométrica, durante el primer mes sólo hubo una mejora del consumo de oxígeno en el grupo de estenosis (24 ± 8 frente a 27 ± 9 ml/kg/min), sin modificaciones al año en ninguno de los dos grupos. En este trabajo se describe que la remodelación aguda del VD que se obtiene con la IPVP, así como la mejoría de su función (sólo en caso de que la estenosis sea la lesión dominante), se mantiene inalterable después de 1 año. A partir de estos resultados, podemos concluir que la implantación percutánea de la válvula pulmonar serviría para detener la dilatación y el deterioro de la función del VD, sin que debamos esperar más mejoría después de 1 año. El tratamiento agresivo y precoz de la estenosis y/o IP posquirúrgicas, sin esperar al deterioro del VD, parecería la alternativa más indicada, siempre que la técnica sea posible. Por otro lado, la experiencia adquirida con ella ha permitido ampliar su utilización a otras posiciones valvulares. Así, Roberts et al40 han presentado la primera serie de implantación de válvula percutánea en posición tricuspídea, con buenos resultados.

Figura 4. Paciente de 9 años con atresia pulmonar, septum integro y prótesis pulmonar biológica de 18 mm implantada a los 2 años de edad. A: angiografía lateral en la que se evidencia insuficiencia severa de la endoprótesis. B: angiografía posterior a la implantación de prótesis percutánea Melody, con desaparición de la insuficiencia.
TRASPLANTE CARDIACO
Han sido muchas las publicaciones aparecidas en los últimos meses respecto a este tema, circunscritas al ámbito de las CC y a la edad pediátrica. Revisamos los trabajos más significativos sobre distintos temas.
Volumen y resultados
Destaca la publicación de Davies et al41 con los datos de la United Network for Organ Sharing sobre 4.647 trasplantados menores de 19 años. Se registra relación entre centros con mayor volumen de pacientes y supervivencia. Proponen la centralización y regionalización de los centros trasplantadores para mejorar los resultados.
Sensibilización
Se han publicado nuevos datos sobre factores de riesgo de mortalidad relacionada con el trasplante. Scott et al42 han presentado una serie de 101 pacientes pediátricos, en los que un Panel Reactive Antibody (PRA) > 80% supone mayor mortalidad e incidencia de rechazos humorales. Mahle et al43 han presentado a 1.904 pacientes con datos de PRA seguidos en el Pediatric Heart Transplant Study Group, con conclusiones parecidas al estudio anterior.
Virus
Breinholt et al44 relacionan la presencia de Parvovirus B19 en el corazón trasplantado y el desarrollo de vasculopatía del injerto. Por otro lado, como describen Snydman et al45, la prevención de citomegalovirus con inmunoglobulina anticitomegaloviral asociada o no con antivirales parece mejorar la supervivencia. La pandemia de gripe A H1N1 produjo grandes debates, especialmente sobre su prevención y sus efectos en pacientes pediátricos de riesgo como los trasplantados de órgano sólido. Vázquez-Álvarez et al46 han descrito la eficacia y la seguridad de la vacunación contra este virus en una población de 67 pacientes con 11 infecciones (tasa, 16,3%). El riesgo relativo de infección aumentó el 2,6% (IC95%, 0,96-9,05) en los no vacunados. Por otro lado, el tratamiento con oseltamivir se toleró bien y la evolución general de los cuadros fue benigna.
Enfermedad linfoproliferativa postrasplante (PTLD)
Gajarski et al47 han analizado el registro multicéntrico de 2.374 pacientes pediátricos, y observan que, con terapias de inducción agresivas (sobre todo OK3), no hay más riesgo de infección fúngica ni de PTLD como se pensaba hasta ahora.
De los números a las guías clínicas
Los cambios demográficos, las nuevas necesidades asistenciales, los déficit de formación de los cardiólogos clínicos y, sobre todo, la ausencia de criterios objetivos en las indicaciones terapéuticas de los adultos con CC, han promovido el desarrollo de nuevas guías de práctica clínica para el manejo de esta población cardiovascular emergente.
En noviembre de 2008 aparecieron las guías clínicas sobre CC del adulto de la American Heart Association; en marzo de 2010 se publicaron las guías canadienses y, por fin, en diciembre de 2010 se publicó la nueva versión de las guías clínicas de la ESC49. Estas nuevas guías mantienen una corta sección sobre los problemas generales, pero se centran en las recomendaciones específicas para el manejo de las diferentes CC que aparecen más frecuentemente en la población adulta, especialmente en pacientes con CC reparadas. Cada lesión específica se analiza en términos de su historia natural y las modificaciones impuestas por las intervenciones terapéuticas, se indica el proceso diagnóstico enfatizando la utilidad de la ecocardiografía y las nuevas herramientas radiológicas, se actualizan los métodos terapéuticos y el timing de las intervenciones, se simplifican las indicaciones quirúrgicas y los procedimientos percutáneos basados en conceptos clínicos y datos fácilmente obtenidos por técnicas de imagen y se establecen las principales recomendaciones en el seguimiento de estos pacientes. La principal limitación de estas guías sigue siendo la ausencia de estudios basados en la evidencia, por lo que la mayoría de las recomendaciones son de tipo C, es decir, que sólo se basan en la opinión de los expertos.
Un problema cada vez más frecuente en el área de la cardiología es el embarazo de mujeres con cardiopatía y, aunque no únicamente dedicadas a las CC, teniendo en cuenta la prevalencia de estas entre las mujeres gestantes con una anomalía cardiaca (75-82%), creemos importante mencionar la publicación (agosto de 2011) auspiciada por la ESC50 de unas guías de práctica clínica sobre ese tema. No obstante, de forma similar a lo indicado en el apartado previo, nuevamente casi todas las recomendaciones son de tipo C, puesto que no se dispone de suficientes estudios que las avalen.
CONFLICTO DE INTERESES
Ninguno.
*Autor para correspondencia: Torrent de la Verdaguera 3, 08110 Montcada-Reixac, Barcelona, España.
Correo electrónico: msubiranad@santpau.cat (M.T. Subirana).