


La enfermedad de Danon (ED) es una enfermedad producida por mutaciones en el gen LAMP2. Se la considera una enfermedad multisistémica caracterizada por miocardiopatía hipertrófica con preexcitación e hipertrofia extrema, discapacidad intelectual, miopatía, presentación infantil y peor pronóstico en varones. Hay pocas series que permitan conocer las características clínicas y el pronóstico de la ED.
MétodosSe analizaron los registros clínicos de los pacientes con ED de 10 hospitales españoles.
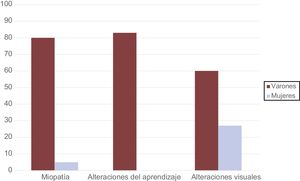
ResultadosSe incluyó a 27 pacientes (edad, 31 ± 19 años; el 78% mujeres). Los varones mostraron una elevada prevalencia de manifestaciones extracardiacas —miopatía (80%), trastornos del aprendizaje (83%) y alteraciones visuales (60%)— que eran infrecuentes en las mujeres (el 5, el 0 y el 27% respectivamente). Aunque la miocardiopatía hipertrófica era la cardiopatía más habitual (61%), el grosor ventricular máximo fue 15 ± 7 mm y 12 pacientes (10 mujeres) presentaron miocardiopatía dilatada. Solo 11 pacientes (49%) mostraron preexcitación y en 16 (65%) la enfermedad se inició después de los 20 años. Tras una mediana de seguimiento de 4 años [intervalo intercuartílico, 2-9], 4 varones (67%) y 9 mujeres (43%) fallecieron o se sometieron a trasplante. El daño cardiaco y los eventos adversos ocurrieron más tardíamente en las mujeres (37 ± 9 frente a 23 ± 16 años y 36 ± 20 frente a 20 ± 11 años).
ConclusionesLas características clínicas de la ED difieren sustancialmente de lo considerado tradicionalmente. La edad de presentación de la ED es más tardía, no se expresa como una enfermedad multisistémica en las mujeres y la preexcitación es poco frecuente.
Palabras clave
La enfermedad de Danon (ED) es muy poco frecuente y está clasificada dentro del grupo de las enfermedades por depósito de glucógeno. Su herencia esá ligada al cromosoma X y se produce por una mutación en el gen LAMP2, que codifica para la proteína asociada con la membrana lisosomal 2 (OMIM# 309060.0005)1. Es la alteración de esta proteína de transporte, y no un déficit en la maltasa ácida como el resto de enfermedades de depósito, lo que hace que se produzca una acumulación sistémica de glucógeno que puede afectar a órganos como el corazón o los músculos. Así pues, el cuadro clínico clásico de la ED descrito por Danon et al.2 en 1981 se caracteriza por miocardiopatía, daño muscular y retraso mental. Asimismo, se considera que la ED conlleva muy mal pronóstico y que suele iniciarse en la infancia o la adolescencia3.
El daño cardiaco típico de la ED consiste en una miocardiopatia hipertrófica (MCH) con grosores ventriculares muy aumentados y preexcitación en el electrocardiograma (ECG)3,4.
La ED está poco caracterizada, hay muy poca información en la literatura en cuanto a clínica, evolución y pronóstico. Además, los datos son especialmente escasos en lo relativo a la población femenina.
Se analizaron los datos procedentes del registro español de pacientes con ED, con el fin de determinar su perfil clínico y su curso evolutivo.
MÉTODOSSe analizaron retrospectivamente la información clínica y las pruebas de 27 pacientes con ED provenientes de 17 familias atendidos en 10 hospitales universitarios españoles. El registro español de ED está abierto a todos los hospitales que deseen participar y se coordina desde el Hospital Universitario Puerta de Hierro Majadahonda, en Madrid. El criterio de inclusión de los pacientes en el registro es que tengan una mutación en el gen LAMP2 confirmada genéticamente y considerada patogénica.
La patogenicidad de las variantes detectadas (tabla 1) se catalogó en función de que existieran descripciones previas, que estuvieran registradas en bases de datos públicas de la población general (Exome Aggregation Consortium, Exome Variant Server y ClinVar) o una base de datos de patogenicidad (Human Genome Mutation Database), la presentación de un fenotipo compatible y la información familiar, de acuerdo con los criterios publicados por el American College of Medical Genetics and Genomics5.
Variantes patogénicas encontradas en LAMP2, frecuencia y distribución por sexo
| Posición genómica | Nivel de proteína (NP_002285.1) | MAFExAC | EVS MAF (%) (EA/AA/todos) | ClinVar | HGMD | n | V (n = 6) | M (n = 21) | Fenotipo |
|---|---|---|---|---|---|---|---|---|---|
| g.119590551C>T | p.Trp46* | NI | NI | NI | CM132566Patogénica | 6 | 3 | 3 | MCHMiopatía |
| g.119576454C>T | p.Val310Ile | NI | NI | rs104894858Patogénica | CM057189Patogénica | 2 | 0 | 2 | MCH |
| g.119580245_119580246insT | p.His260GInfs*14 | NI | NI | NI | NI | 3 | 0 | 3 | MCH |
| ChrX:119573044-119636423del [ChrX:119635816-119635989]ins | NI | NI | NI | NI | 2 | 0 | 2 | MCH | |
| g.119580246G>A | p.His260Tyr | 2/87.696(0,0022) | NI | rs778577575Significado incierto | NI | 2 | 1 | 1 | MCHPreexcitación |
| g.119575626delC | p.Val352Phefs*6 | NI | NI | NI | NI | 2 | 0 | 2 | MCD |
| g.119589350_119589351insT | p.Ile88Asnfs*25 | NI | NI | NI | NI | 1 | 0 | 1 | MCD |
| g.119582963delG | p.Leu140Phefs*8 | NI | NI | NI | NI | 1 | 0 | 1 | MCHPreexcitación |
| g.119590506delA | p.Tyr61* | NI | NI | NI | NI | 2 | 1 | 1 | MCD |
| g.119576505G>A | p.Arg293* | NI | NI | rs727503118Patogénica | HM070116Patogénica | 2 | 0 | 2 | MCDPreexcitación |
| g.119582962delA | p.Leu141Trpfs*7 | NI | NI | NI | NI | 1 | 0 | 1 | MCH |
| g.119581726delC | p.Leu239Cysfs*3 | NI | NI | NI | NI | 1 | 0 | 1 | MCD |
| g.119576454C>A | p.Val310Phe | NI | NI | NI | NI | 1 | 0 | 1 | MCH |
| g.119575602_119575603insT | p.Gly360Argfs*13 | NI | NI | NI | NI | 1 | 1 | 0 | MCH |
AA: African American; ClinVar: Clinical Variant; EA: European American; ExAC: Exome Aggregation Consortium; EVS: Exome Variant Server; HGMD: Human Genome Mutation Database; M: mujeres; MAF: Minor allele frequency; MCD: miocardiopatía dilatada; MCH: miocardiopatía hipertrófica; NI: no identificado; V: varones.
La información de cada paciente se obtuvo a través de una hoja de recogida de datos estandarizada común. Para cumplimentarla, se realizó una revisión exhaustiva de los registros clínicos, tanto en papel como en soporte electrónico.
Análisis estadísticoLos resultados numéricos se expresaron como media ± desviación estándar, en el caso de las variables numéricas continuas con distribución normal, y como mediana (máximo-mínimo), para las continuas no normales, o porcentajes, para las categóricas. Para las comparaciones entre sexos, se utilizaron pruebas de la t de Student bilaterales para muestras independientes en las variables continuas de distribución normal, pruebas no paramétricas para las no normales y el test exacto de Fisher para las variables binarias. El nivel de significación se estableció en p < 0,05. El tratamiento estadístico se llevó a cabo mediante el paquete SPSS versión 20 (IBM Corp.; Amronk, Nueva York, Estados Unidos).
RESULTADOSSe analizaron los datos de una cohorte de 27 pacientes con mutaciones patogénicas en el gen LAMP2, solo el 22% de los individuos eran varones (n = 6); 17 pacientes (63%) fueron los primeros diagnosticados en sus familias (casos índice). Se diagnosticó a los 10 restantes en el transcurso de la evaluación familiar tras el diagnóstico de ED en otro de los integrantes de sus familias. Solo 5 de los pacientes (4 mujeres) eran portadores de variantes missense (tabla 1). En lo que se refiere a la variante g.119580246G>A, indicaban su patogenicidad un fenotipo compatible y los familiares afectados.
En total, 21 pacientes mostraron algún tipo de daño de la enfermedad en el momento del diagnóstico de ED, 2 lo contrajeron durante el seguimiento y 4 (3 mujeres y 1 varón) eran portadores sin signos de daño en el seguimiento. En 2 individuos el diagnóstico de la enfermedad se produjo en el transcurso de su autopsia tras su fallecimiento (autopsia molecular).
La media de edad en el momento del diagnóstico fue 31 ± 19 años. El diagnóstico de los varones, por lo general, tuvo lugar a una edad más precoz que el de las mujeres (14 ± 7 frente a 35 ± 19 años), con diferencia estadísticamente significativa (p = 0,02) (tabla 2).
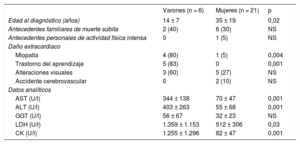
Características clínicas y hallazgos analíticos del daño extracardiaco en pacientes con enfermedad de Danon por sexo
| Varones (n = 6) | Mujeres (n = 21) | p | |
|---|---|---|---|
| Edad al diagnóstico (años) | 14 ± 7 | 35 ± 19 | 0,02 |
| Antecedentes familiares de muerte súbita | 2 (40) | 6 (30) | NS |
| Antecedentes personales de actividad física intensa | 0 | 1 (5) | NS |
| Daño extracardiaco | |||
| Miopatía | 4 (80) | 1 (5) | 0,004 |
| Trastorno del aprendizaje | 5 (83) | 0 | 0,001 |
| Alteraciones visuales | 3 (60) | 5 (27) | NS |
| Accidente cerebrovascular | 0 | 2 (10) | NS |
| Datos analíticos | |||
| AST (U/l) | 344 ± 138 | 70 ± 47 | 0,001 |
| ALT (U/l) | 403 ± 263 | 55 ± 68 | 0,001 |
| GGT (U/l) | 56 ± 67 | 32 ± 23 | NS |
| LDH (U/l) | 1.359 ± 1.153 | 512 ± 306 | 0,03 |
| CK (U/l) | 1.255 ± 1.296 | 82 ± 47 | 0,001 |
ALT: alanina aminotransferasa; AST: aspartato aminotransferasa; CK: creatincinasa; GGT: gamma glutamil transpeptidasa; LDH: lactato deshidrogenasa; NS: no significativo.
Los valores expresan n (%) o media ± desviación estándar.
En 8 pacientes se observaron antecedentes familiares de muerte súbita (un 29% de toda la cohorte).
Daño multisistémicoEn lo que se refiere al daño extracardiaco, las mujeres solo presentaron manifestaciones de daño muscular, visual, hepático o del nivel de aprendizaje de manera esporádica (tabla 2 y figura 1). Así pues, mientras que el 80% de los varones presentaban daño muscular, se observó en solo el 5% de las mujeres. Estos datos se correlacionan con el hallazgo de valores de creatincinasa en sangre significativamente mayores en los varones que en las mujeres (1.255 ± 1.296 frente a 82 ± 47 U/l; p < 0,05). Los únicos 5 individuos de la cohorte que presentaban un trastorno del aprendizaje eran varones (el 83% de la población masculina). La afección en estos casos se demostraba como un retraso mental leve-moderado, a pesar de lo cual, todos los individuos eran totalmente autónomos para las actividades de la vida diaria.

La elevación de enzimas hepáticas fue también un rasgo que se encontró más frecuentemente en la población masculina, con valores medios de AST (aspartato aminotransferasa) y ALT (alanina aminotransferasa) superiores en los varones que en las mujeres (tabla 2).
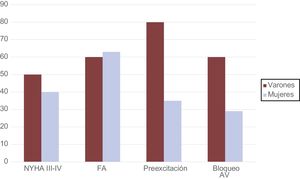
Daño cardiacoLos síntomas a la presentación clínica de los pacientes con ED descritos fueron síncope, dolor torácico y síntomas de insuficiencia cardiaca (clase funcional de la New York Heart Association III-IV) en el 22, el 22 y el 43% de los afectados respectivamente (tabla 3).
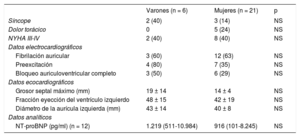
Datos del daño cardiaco en pacientes con enfermedad de Danon por sexo
| Varones (n = 6) | Mujeres (n = 21) | p | |
|---|---|---|---|
| Síncope | 2 (40) | 3 (14) | NS |
| Dolor torácico | 0 | 5 (24) | NS |
| NYHA III-IV | 2 (40) | 8 (40) | NS |
| Datos electrocardiográficos | |||
| Fibrilación auricular | 3 (60) | 12 (63) | NS |
| Preexcitación | 4 (80) | 7 (35) | NS |
| Bloqueo auriculoventricular completo | 3 (50) | 6 (29) | NS |
| Datos ecocardiográficos | |||
| Grosor septal máximo (mm) | 19 ± 14 | 14 ± 4 | NS |
| Fracción eyección del ventrículo izquierdo | 48 ± 15 | 42 ± 19 | NS |
| Diámetro de la aurícula izquierda (mm) | 43 ± 14 | 40 ± 8 | NS |
| Datos analíticos | |||
| NT-proBNP (pg/ml) (n = 12) | 1.219 (511-10.984) | 916 (101-8.245) | NS |
NS: no significativo; NT-proBNP: fracción aminoterminal del propéptido natriurético cerebral; NYHA: New York Heart Association.
Los valores expresan n (%) o media ± desviación estándar.
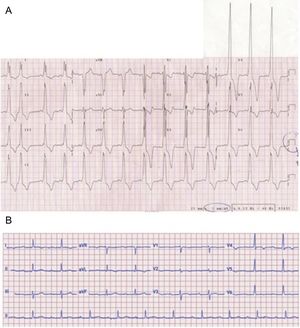
En la revisión sistemática de los ECG de los pacientes con ED incluidos en el registro, solo se documentó un patrón de preexcitación en 11 individuos (48%), 4 de ellos varones (figura 2). La presencia de fibrilación auricular fue muy habitual, y más de la mitad tanto de varones como de mujeres presentaban esta arritmia (figura 3). Además, cabe resaltar que, mientras 4 mujeres presentaban taquicardia ventricular sostenida en el seguimiento (1 de ellas degeneró en fibrilación ventricular), ninguno de los varones sufrió alguna de estas arritmias.


Hallazgos electrocardiográficos en 2 pacientes con enfermedad de Danon. A: ECG de un varón de 14 años con gran hipertrofia ventricular y preexcitación; obsérvese que el ECG se realizó a mitad de voltaje. B: ECG de un varón de 23 años, sin preexcitación ni signos de hipertrofia ventricular. ECG: electrocardiograma.
En nuestra serie, 11 pacientes (41%), 8 mujeres y 3 varones, recibieron el implante de un desfibrilador automático implantable (DAI). Solo en 1 caso la indicación fue por prevención secundaria para una mujer con un claro fenotipo (MCH y preexcitación) que sufrió una taquicardia ventricular sostenida. Los implantes en prevención primaria se realizaron con base en los antecedentes de muerte súbita en la familia y/o disfunción ventricular. Durante el seguimiento, 1 individuo recibió descargas apropiadas del dispositivo y otro, terapias para el tratamiento de una taquicardia ventricular sostenida aparecida en el contexto de tratamiento inotrópico con levosimendán. En ningún otro paciente con DAI se detectaron arritmias ventriculares.
La aparición de trastornos de la conducción también fue habitual, y el 50% de los varones (n = 3) y el 29% de las mujeres (n = 6) sufrieron bloqueo auriculoventricular completo, sin diferencias estadísticamente significativas entre los subgrupos.
Todos los pacientes testados (n = 12) tenían concentraciones aumentadas de la fracción aminoterminal del propéptido natriurético cerebral de manera generalizada, con tendencia a ser mayores en los varones que en las mujeres: 1.219 (511-10.984) frente a 916 (101-8.245) pg/ml (p > 0,05).
En lo referente a los hallazgos ecocardiográficos, el grosor medio del ventrículo izquierdo fue 15 ± 7mm, con una media de fracción de eyección del ventrículo izquierdo (FEVI) de toda la cohorte moderadamente deprimida (43 ± 18%). Es interesante que, en 12 de los sujetos incluidos en el registro (40%), la miocardiopatía que presentaban en el momento del diagnóstico era miocardiopatía dilatada (MCD), con una media de FEVI del 27 ± 8%. Por sexos, las mujeres tuvieron tendencia a mostrar valores de grosor ventricular inferiores (14 ± 4 frente a 19 ± 14mm; p = 0,21) y también menor FEVI (el 42 ± 19% frente al 48 ± 15%; p = 0,49). El 48% de las mujeres frente al 33% de los varones tenían MCD en el momento del diagnóstico (p = 0,54). En lo que se refiere a diferencias según el tipo de mutación, no se detectaron diferencias estadísticamente significativas en las características registradas. Sin embargo, los portadores de una variante missense (5 pacientes) presentaban un fenotipo mayoritariamente en el espectro de la MCH con FEVI conservada. En ellos se observó además escaso daño multisistémico por parámetros tanto clínicos como analíticos (tabla 4).
Datos del daño cardiaco en pacientes con enfermedad de Danon por tipo de mutación
| Missense (n = 5) | No missense (n = 22) | p | |
|---|---|---|---|
| Edad al diagnóstico (años) | 30 ± 16 | 31 ± 4 | NS |
| Daño extracardiaco | |||
| Miopatía | 0 | 5 (23) | NS |
| Trastorno del aprendizaje | 0 | 5 (23) | NS |
| Alteraciones visuales | 1 (20) | 7 (32) | NS |
| Datos analíticos | |||
| AST (U/l) | 84 ± 17 | 129 ± 33 | NS |
| ALT (U/l) | 41 ± 12 | 135 ± 46 | NS |
| GGT (U/l) | 18 ± 3 | 40 ± 9 | NS |
| LDH (U/l) | 923 | 710 ± 184 | NS |
| CK (U/l) | 114 ± 34 | 352 ± 185 | NS |
| Daño cardiaco | |||
| Grosor septal máximo (mm) | 17 ± 4 | 15 ± 2 | NS |
| Fracción de eyección del ventrículo izquierdo | 58 ± 5 | 40 ± 4 | NS |
| Preexcitación | 1 (20) | 10 (46) | NS |
| NYHA III-IV | 1 (20) | 9 (41) | NS |
ALT: alanina aminotransferasa; AST: aspartato aminotransferasa; CK: creatincinasa; GGT: gamma glutamil transpeptidasa; LDH: lactato deshidrogenasa; NS: no significativo; NYHA: New York Heart Association.
Los valores expresan n (%) o media ± desviación estándar.
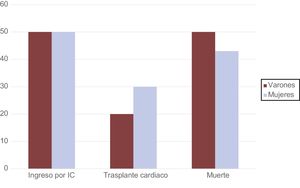
La mediana de seguimiento de los pacientes no diagnosticados tras el estudio de su autopsia fue de 4 años [intervalo intercuartílico, 2-9]. Durante dicho periodo, un 48% de los individuos con ED ingresó a causa de insuficiencia cardiaca (el 50% de los varones y el 47% de las mujeres) (tabla 5). La media de edad cuando ocurrió el primer episodio de insuficiencia cardiaca fue de 23 ± 16 años en los varones y 37 ± 9 años en las mujeres (p = 0,09).
Eventos y edad de aparición de estos en pacientes con enfermedad de Danon por sexo
| Varones (n = 6) | Mujeres (n = 21) | p | |
|---|---|---|---|
| Ingreso por insuficiencia cardiaca | 2 (50) | 10 (47) | NS |
| Edad en el primer episodio de insuficiencia cardiaca (años) | 23 ± 16 | 37 ± 9 | NS |
| Trasplante cardiaco | 1 (20) | 6 (30) | NS |
| Edad al trasplante cardiaco (años) (n = 7) | 14 | 32 ± 9 | NS |
| Muerte | 3 (50) | 3 (14) | NS |
| Edad al fallecimiento (años) (n = 6) | 23 ± 13 | 40 ± 14 | NS |
| Muerte o trasplante cardiaco | 4 (57) | 9 (43) | NS |
| Edad a la muerte o al trasplante cardiaco (años) (n = 13) | 20 ± 11 | 36 ± 20 | NS |
NS: no significativo.
Los valores expresan n (%) o media ± desviación estándar.
En nuestra cohorte total, se registraron en total 6 fallecimientos (el 22% de la población) (figura 4). De todos los fallecimientos, 3 eran varones (50%): 1 por insuficiencia cardiaca y 2 por muerte súbita. De las 3 mujeres fallecidas, la causa de muerte de 2 de ellas fue la insuficiencia cardiaca avanzada y la de la restante, muerte súbita. Además, 7 individuos recibieron trasplante cardiaco; 1 era varón. El trasplante cardiaco tuvo lugar en el varón a la edad de 14 años, mientras que para las mujeres fue a una media de 32 ± 9 (18-43) años.

Al igual que en lo relativo a la insuficiencia cardiaca, también se encontró que la media de edad al trasplante o el fallecimiento fue superior en las mujeres que en los varones, aunque la diferencia no era significativa (36 ± 20 frente a 20 ± 11 años; p = 0,14) (figura 4).
DISCUSIÓNEn este trabajo se recogen las características clínicas y el curso clínico de una de las cohortes más numerosas de pacientes con ED descritas hasta la fecha. El análisis de los pacientes con ED recogidos en nuestro trabajo mostró que las características clínicas de la ED difieren sustancialmente de lo tradicionalmente considerado en esta enfermedad. Nuestros datos muestran que la ED no se expresa habitualmente como una enfermedad multisistémica en las mujeres, que la preexcitación es un hallazgo electrocardiográfico poco frecuente en los pacientes con ED y que la edad de presentación habitual de los pacientes con ED es más tardía que lo descrito hasta la fecha. Además, nuestros datos confirman que, aunque las mujeres con ED tienen un pronóstico clínico desfavorable, los eventos adversos en ellas ocurren a una edad sustancialmente más avanzada que en los varones.
La ED es una enfermedad muy poco frecuente ligada al cromosoma X. Se produce por mutaciones en el gen LAMP2, que causan un déficit total o parcial de la actividad de la proteína LAMP-2. El déficit de actividad de esta enzima conduce a la acumulación de glucógeno en el interior de los lisosomas, lo que constituye el sustrato histopatológico de la ED. Hasta la fecha se han descrito más de 100 mutaciones diferentes en LAMP2 que dan lugar a ED6.
La ED se considera una enfermedad multisistémica que se caracteriza por la tríada clásica de discapacidad intelectual, miopatía y MCH con hipertrofia ventricular extrema y preexcitacion en el ECG3,4. Además, en los pacientes con ED pueden concurrir también, aunque con menos frecuencia, retinopatía y enfermedad pulmonar o hepática2, daño escasamente descrito en nuestros pacientes. En general, y dado el contexto de herencia ligada al cromosoma X, tal y como se observó en nuestra serie, los varones suelen presentar un cuadro clínico más grave y de aparición más precoz que las mujeres, comenzando con síntomas y signos de la enfermedad en la infancia o en la adolescencia3. En cambio, en el caso de las mujeres, su condición de heterocigosis para la mutación hace que se considere poco frecuente la aparición de síntomas antes del inicio de la edad adulta7.
La prevalencia exacta de la ED se desconoce, pero publicaciones recientes apuntan a que es mayor que la previamente descrita y se puede alcanzar el 1-6% de los pacientes con MCH de etiología desconocida4,8. Esta prevalencia puede aumentar hasta el 17% si se selecciona a los pacientes con MCH junto con otras características, como la elevación de la creatincinasa o el patrón de preexcitación en el ECG4.
Aunque el daño muscular en los pacientes con ED se manifiesta como debilidad, intolerancia al ejercicio y aumento de la creatincinasa, esta tríada se encuentra en nuestra población casi exclusivamente en los varones y solo 1 de las 21 mujeres incluidas en nuestro registro se aquejaba de dolencias musculares.
Se encontraron alteraciones del aprendizaje en solo un 19% de los pacientes con ED, valores mucho más bajos que los descritos clásicamente en la literatura (un 70-100% de los varones y hasta el 47% de las mujeres)6. En concordancia con nuestro hallazgo, un estudio muy reciente en el que se realizó una evaluación psicológica y cognitiva detallada mostró que las alteraciones cognitivas y psiquiátricas no son tan comunes en los pacientes con ED9. En cambio, en este estudio reciente se destaca que los trastornos del ánimo y la ansiedad son frecuentes en los pacientes con ED y que podrían afectar a un 70%, y con igual afección en ambos sexos9.
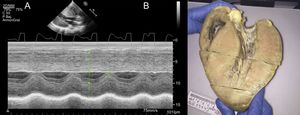
La manifestación cardiaca de la ED más comúnmente descrita es la MCH con FEVI conservada. Es por este motivo que la ED se considera una fenocopia de la MCH10, la cual, además, suele tener un peor pronóstico que la MCH causada por mutaciones sarcoméricas8,11. La hipertrofia cardiaca de los pacientes con ED puede ser extrema (figura 5, varón de 14 años trasplantado en nuestra cohorte) y, de hecho, el corazón más grande publicado hasta la fecha es el de un paciente americano de 14 años con ED, cuya autopsia reveló que tenía un grosor de 65mm y un peso de 1.425g3.
 y macroscópica (B) del corazón de un varón de 14 años con enfermedad de Danon e hipertrofia del ventrículo izquierdo masiva (septo interventricular de 43 mm). Imagen macroscópica cortesía de la Dra. Elena Ruiz del Hospital La Paz.")
Además del daño cardiaco en forma de MCH, es relevante destacar que un subgrupo de pacientes con ED lo presenta en forma de MCD. Este grupo se compone particularmente por mujeres y en nuestra cohorte alcanza el 41% de las mujeres con algún tipo de miocardiopatía. Por eso no se debe descartar la ED en caso de que se demuestre MCD, y particularmente se recomienda sospecharla en individuos con MCH que evolucione a forma dilatada9,12. Además es recomendable incluir el gen LAMP2 en los paneles de genes que se emplean en los estudios genéticos de sujetos con MCD y no reservarlo únicamente para pacientes con MCH10.
En pacientes diagnosticados de ED son comunes las alteraciones de la conducción y las arritmias. El patrón de preexcitación en el ECG se ha descrito en un 68% de los varones y un 27% de las mujeres7 y constituye un importante signo que permite sospechar la ED10. Sin embargo, nuestra serie muestra que la preexcitación es menos frecuente de lo que se ha publicado tradicionalmente y solo la mostraban el 41% de los pacientes, lo que pudo dificultar el diagnóstico en algunos casos.
En cuanto a las arritmias, diversos trabajos previos con un número de sujetos menor que en nuestra serie comunicaron una alta tasa de arritmias ventriculares, particularmente entre los varones con ED. En nuestra serie, 2 varones y 1 mujer con ED fallecieron súbitamente (ninguno portador de DAI) y otros 4 (todas mujeres) presentaban taquicardia ventricular sostenida al seguimiento. La tasa de arritmias graves/muerte súbita en nuestra cohorte es alta (el 26% de los sujetos) y superior a la indicada en publicaciones recientes cuya tasa de arritmias ventriculares/muerte súbita se sitúa en torno al 14%13.
Los datos de nuestro registro confirman que la ED tiene mal pronóstico en ambos sexos, y en ausencia de trasplante cardiaco los varones no alcanzan los 25 años de vida. Asimismo, hay que destacar que se han descrito diversos casos de descargas de DAI que no consiguieron la terminación de las arritmias ventriculares en algunos sujetos con ED3.
Por ello el diagnóstico precoz de la enfermedad, basado en un alto nivel de sospecha, resulta de vital importancia, ya que es necesario establecer una estrategia terapéutica y un plan de cuidados que permitan adelantarse a las posibles complicaciones que pueden presentarse en estos pacientes.
Actualmente no hay guías específicas para el tratamiento de la ED y tampoco se han llevado a cabo ensayos clínicos en este campo. De hecho, las indicaciones tanto de DAI como de trasplante son similares a las establecidas para la MCH y la MCD. Así pues, en los casos que presenten un fenotipo de MCH, la indicación se establecerá con base en los factores de riesgo clásicos de muerte súbita y MCH. En cambio, si este es de MCD, son la FEVI y la clase funcional lo que indique el implante de DAI. Recientemente se publicaron una serie de recomendaciones para profesionales sobre el seguimiento y el tratamiento de la ED14. Uno de los aspectos más relevantes que destacar es que el tratamiento de los pacientes con ED necesita un abordaje multidisciplinario. El equipo médico ha de reunir a cardiólogos, neurólogos, oftalmólogos, psiquiatras y genetistas clínicos.
LimitacionesAunque nuestro trabajo recoge una de las series más extensas de ED descritas hasta la fecha, hay que reconocer que incluye un número de sujetos pequeño. Asimismo, los datos se recogieron de manera retrospectiva mediante revisión de las historias clínicas de los pacientes, con los sesgos de información, de supervivencia o de selección que esto puede suponer. En este contexto, para la evaluación de ciertos parámetros como las alteraciones del aprendizaje, no se emplearon protocolos específicos de estudio. Es importante destacar el pequeño número de varones incluidos y las posibles consecuencias en el análisis estadístico que esto conlleva. Además, el hecho de tratarse de un registro en el que participan numerosos centros de referencia puede hacer que haya un sesgo de selección por infrarrepresentación de casos con manifestaciones clínicas más leves.
CONCLUSIONESLa ED tiene mal pronóstico, la cual se vincula al daño cardiaco. Este se produce en sujetos de ambos sexos, pero característicamente aparece más tardíamente en las mujeres.
La ausencia de la tríada clásica no permite descartar la enfermedad, sobre todo en las mujeres, puesto que en ellas el daño puede ser exclusivamente cardiaco. Además, signos característicos considerados típicos, como la preexcitación en el ECG, pueden aparecer solo en una minoría de los individuos con ED.
El conocimiento más en profundidad de las características clínicas y los mecanismos fisiopatológicos de la ED facilitará el diagnóstico de esta entidad, para la que es importante establecer una estrategia apropiada de tratamiento que incluya la consideración precoz para trasplante cardiaco.
FINANCIACIÓNEste trabajo se ha realizado en parte gracias a ayudas del Instituto de Salud Carlos III (PI14/01477 y La Fe Biobank PT17/0015/0043), que cuentan con financiación de fondos FEDER «Una forma de hacer Europa».
CONFLICTO DE INTERESESNo se declara ninguno.
- –
La ED es una enfermedad poco frecuente ligada al cromosoma X y producida por mutaciones en el gen LAMP2, se la considera una enfermedad multisistémica caracterizada por MCH con preexcitación y gran hipertrofia, discapacidad intelectual, miopatía, presentación infantil y peor pronóstico en los varones.
- –
Se trata de una enfermedad mal caracterizada, con muy pocas series publicadas en la literatura, por lo que hay muy poca información en lo que a clínica, evolución y pronóstico se refiere. Además, los datos son especialmente escasos en lo relativo a la población femenina.
- –
Las características clínicas de la ED difieren sustancialmente de lo tradicionalmente considerado. Según nuestros datos, la ED no se expresa como una enfermedad multisistémica en las mujeres y la edad de presentación en ellas es más tardía. La preexcitación en el ECG aparece en una minoría de pacientes. Siendo el daño cardiaco lo que marca el pronóstico, en ausencia de trasplante cardiaco los varones no alcanzan los 25 años de vida. Aunque las mujeres presentan también mala evolución, los eventos adversos aparecen a una edad más avanzada. A la vista de nuestros datos, la ED podría considerarse fenocopia tanto de MCH como de MCD, puesto que un porcentaje no desdeñable (especialmente de mujeres) se inicia directamente como MCD.
Al Dr. Herminio Morillas por la remisión de pacientes para este estudio y a la Dra. Elena Ruiz por facilitarnos la imagen macroscópica de la figura 5.