
Los ensayos clínicos aleatorizados (ECA) de no inferioridad se suelen llevar a cabo en circunstancias en que no es esperable que el tratamiento experimental sea más eficaz que el tratamiento estándar, pero el nuevo fármaco ofrece ventajas adicionales. Dichas ventajas podrían ser un mejor perfil de seguridad, menos efectos secundarios, más fácil administración, menos necesidad de controles o un menor coste total1. Los ensayos RE-LY (Randomized Evaluation of Long-Term Anticoagulation Therapy), ROCKET-AF (Rivaroxaban Once Daily Oral Direct Factor Xa Inhibition Compared With Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation) y ARISTOTLE (Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation) son ECA de no inferioridad frente a la warfarina. Seguidamente se repasan varios conceptos relevantes para la interpretación de dichos estudios, que son aplicables en general a todos los ECA de no inferioridad.
¿QUÉ SON LOS ENSAYOS CLÍNICOS DE NO INFERIORIDAD?En los ECA se pretende responder a una pregunta de investigación con un grado razonable de certidumbre. Mientras los ECA de superioridad tratan de probar si un nuevo tratamiento es superior al mejor tratamiento disponible, en los ECA de no inferioridad el esfuerzo se concentra en demostrar que el nuevo tratamiento no es inferior al estándar. La naturaleza de la pregunta de investigación y la de las posibles respuestas son diferentes. En el caso concreto de los ECA RE-LY, ROCKET-AF y ARISTOTLE, la pregunta inicial de interés fue: ¿ el nuevo tratamiento es al menos tan eficaz como la warfarina para la reducción de eventos tromboembólicos? Las dos posibles respuestas, mutuamente excluyentes y formuladas en forma de hipótesis, son:
- •
H0 (hipótesis nula): el nuevo tratamiento es menos eficaz que los antagonistas de la vitamina K para la reducción de eventos tromboembólicos (es inferior).
- •
H1 (hipótesis alternativa): el nuevo tratamiento es al menos tan eficaz como los antagonistas de la vitamina K para la reducción de eventos tromboembólicos (no es inferior).
Adoptar «la respuesta» H0 o H1 como verdadera se fundamenta en una regla de decisión según la significación estadística del valor de p. Sin embargo, el valor de p que se calcula en los ECA de no inferioridad es especial; se denomina p de no inferioridad. Supongamos que la tasa de eventos tromboembólicos con el nuevo tratamiento es inferior que con la warfarina con un valor de p de no inferioridad < 0,001. En este caso, adoptaremos la hipótesis alternativa H1 como cierta, porque si realmente el nuevo tratamiento fuera inferior a los antagonistas de la vitamina K, obtener dicho resultado habría sido tan improbable como p < 0,001.
En los ECA de no inferioridad, hay que definir a priori qué consideramos «al menos tan eficaz» o «no inferior» que el tratamiento convencional. Para ello se ha de seleccionar un margen o dintel mínimo de no inferioridad. El objetivo de los ECA de no inferioridad es demostrar que el efecto del tratamiento experimental, comparado con el del tratamiento estándar, no es inferior a «cierta cantidad», que se denomina dintel mínimo de no inferioridad o margen de no inferioridad o delta (δ). El δ expresa cuán superior se permite como máximo que sea el efecto de la terapia control respecto al tratamiento experimental y que a este se lo pueda seguir considerando no inferior respecto al control2.
¿CÓMO SE ELIGE EL DINTEL MÍNIMO DE NO INFERIORIDAD δ?El margen de no inferioridad δ se ha de seleccionar de acuerdo con la mejor evidencia disponible de la eficacia del tratamiento estándar respecto al placebo2. Para su selección se ha de tener en cuenta el grado de certeza o incerteza que se tenga sobre el efecto del tratamiento estándar, tendiendo a ser conservadores. El margen de no inferioridad no puede exceder el efecto de magnitud más pequeña que sería plausible que tuviera el tratamiento estándar, en nuestro caso la warfarina, con respecto a placebo.
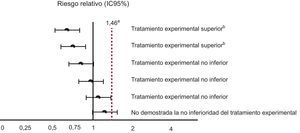
En el caso de los nuevos anticoagulantes, el dintel mínimo de no inferioridad se fijó a partir de un metanálisis publicado en 1999, en el que se cuantificó el efecto de la warfarina en la prevención de eventos tromboembólicos respecto al placebo o la ausencia de tratamiento, en un riesgo relativo (RR) de 0,38 (intervalo de confianza del 95% [IC95%], 0,28-0,52)3. El procedimiento es el siguiente: primero se cambia la categoría de referencia, como si se calculara el efecto del «placebo o ausencia de tratamiento» respecto a la warfarina. En nuestro caso, dicho efecto sería el inverso de 0,38, que corresponde a RR = 2,63 (IC95%, 1,92-3,57). El margen inferior de dicho intervalo de confianza, 1,92, podría considerarse el dintel mínimo de no inferioridad para los nuevos anticoagulantes. Sin embargo, las agencias reguladoras fueron más exigentes, y eligieron un dintel de no inferioridad asumiendo que, hipotéticamente, la warfarina tuviera únicamente un 50% del efecto que realmente tiene. Así, el dintel mínimo de no inferioridad quedaba fijado en 1,46. Es decir, para tomar la decisión de que el nuevo tratamiento no era inferior al estándar, el límite superior del IC95% del efecto del nuevo tratamiento frente a la warfarina no podría superar 1,46. La figura 1 esquematiza los posibles escenarios que se podría obtener en la comparación de los nuevos anticoagulantes frente a warfarina.
¿TIENE ALGUNA PARTICULARIDAD EL ANÁLISIS ESTADÍSTICO EN LOS ENSAYOS CLÍNICOS ALEATORIZADOS DE NO INFERIORIDAD?
El análisis estadístico en los ECA de no inferioridad suele seguir una metodología similar que con los ECA de superioridad, con la particularidad de que se calcula un valor de p de «no inferioridad», en relación con el dintel de no inferioridad, que diferirá del valor de p «de superioridad». Así, por ejemplo, en el ROCKET-AF el efecto del tratamiento en la variable de resultado primaria «ictus o embolia sistémica» en el análisis por intención de tratar (ITT, por sus siglas en inglés) fue hazard ratio (HR) = 0,88 (IC95%, 0,75-1,03). El valor de p de superioridad fue p = 0,12, mientras que el de no inferioridad fue p < 0,001. Lógico si consideramos que el dintel de no inferioridad se encuentra a la derecha del dintel de ausencia de efecto.
Un punto importante para la interpretación de los resultados de los ECA de no inferioridad es la consideración del tipo de análisis llevado a cabo: análisis ITT, análisis por protocolo o análisis de seguridad. En los ITT se analiza a todos los pacientes aleatorizados a una u otra rama del tratamiento, independientemente de si recibieron finalmente el tratamiento o de si hubo o no violaciones de protocolo. El análisis de seguridad incluye a los pacientes que recibieron al menos una dosis del tratamiento, independiente de si hubo o no violaciones del protocolo. El análisis por protocolo incluye a los pacientes que recibieron al menos una dosis del tratamiento sin violaciones del protocolo detectadas. Mientras que en los ECA de superioridad las agencias reguladoras consideran obligado el análisis ITT, en una hipótesis de no inferioridad se considera necesario sobre todo demostrar no inferioridad en el análisis por protocolo o de seguridad2. En los estudios de no inferioridad, el análisis ITT suele ser más «liberal», es decir, la inclusión de pacientes con violaciones de protocolo o que interrumpieron los tratamientos tiende a sesgar los resultados hacia la ausencia de diferencias entre ambos tratamientos, por lo que será más fácil demostrar no inferioridad.
Finalmente, ¿se puede poner a prueba hipótesis de no inferioridad y de superioridad en un mismo ECA? Sí, siempre que se controle el riesgo α, es decir, la probabilidad de rechazar la H0 cuando en realidad esta es verdadera. Cuantas más hipótesis se ponga a prueba en un estudio con los mismos datos, más probable es encontrar una asociación estadística por azar, por lo que, para evitar falsos positivos, se exige «repartir» el error α entre las hipótesis, lo que da lugar a que, para encontrar resultados estadísticamente significativos, el valor de p haya de ser menor. En los tres estudios se realizaron los oportunos ajustes del riesgo α. El análisis del ROCKET-AF tiene, además, ciertas particularidades.
PARTICULARIDADES DEL PLAN DE ANÁLISIS DEL ROCKET-AFEn el ROCKET-AF se realizó un análisis ITT, un análisis de seguridad y un análisis por protocolo para el evento principal (ictus o embolia periférica), con una particularidad: tanto en el análisis por protocolo como en el de seguridad se incluyó únicamente el periodo durante el cual el paciente estaba recibiendo el fármaco experimental o el placebo y hasta 48 h después de haberlo abandonado (as treated). Esto es importante, ya que el método de análisis de supervivencia considera el «tiempo hasta el evento», y no la proporción de eventos. Así, si un paciente estuvo 730 días (2 años) con la medicación en estudio (experimental o placebo) (figura 2) y posteriormente se le hizo seguimiento 90 días más (0,25 años aproximadamente), contribuirá en el análisis por protocolo y de seguridad 732 días (730 días con la medicación + 2 días tras el abandono, ≈2 pacientes-año), mientras que en el análisis ITT contribuirá 820 días (≈2,25 pacientes-año).

El análisis principal de no inferioridad del ROCKET-AF se realizó en la población por protocolo y por ITT. Además, se realizó en la población un análisis de superioridad en seguridad y varios análisis de sensibilidad para valorar no inferioridad y superioridad en la población por ITT, con los oportunos ajustes del riesgo α. Como la hipótesis principal es de no inferioridad, es oportuno el análisis principal en la población por protocolo2. Por otro lado, como admiten reconocidas agencias internacionales y de nuestro entorno, para la evaluación de la efectividad clínica, es adecuado el análisis en la población de seguridad, en la que no se incluye a los pacientes que no llegaron a recibir el tratamiento experimental o que durante el estudio cambiaron al tratamiento control. En el ROCKET-AF se valoró en dicha población de seguridad y en la población por ITT la hipótesis de superioridad (para el evento principal: población de seguridad, HR = 0,79; IC95%, 0,65-0,95; p de superioridad = 0,02; población ITT, HR = 0,88; IC95%, 0,75-1,03; p de superioridad = 0,12)4,5.
¿SON COMPARABLES LOS RESULTADOS DE LOS TRES ESTUDIOS?Como los tres estudios evalúan la misma hipótesis de no inferioridad y todos ellos utilizan warfarina como control, resulta tentador comparar sus resultados. Sin embargo, cualquier comparación que se haga entre ellos será una comparación indirecta. Es decir, las diferencias en las poblaciones de estudio, la intervención control o el diseño son potenciales fuentes de sesgo6. Así, hubo diferencias en el riesgo de tromboembolia: el riesgo de tromboembolia medio medido por CHADS2 fue 3,47 en el ROCKET-AF frente a 2,1 y 2,1 en RE-LY y ARISTOTLE respectivamente. El riesgo de tromboembolia superior fue, fundamentalmente, por la mayor inclusión de pacientes con antecedentes de ictus (el 55% en el ROCKET frente al 20% en el RE-LY y el ARISTOTLE). Además, el tiempo medio del INR (razón internacional normalizada) en intervalo terapéutico en el ROCKET-AF también difirió sustancialmente de los otros estudios (el 55 frente al 65 y el 62,2% respectivamente). Aunque se han desarrollado diversas técnicas analíticas para la realización de comparaciones indirectas —como el metanálisis en red, las comparaciones indirectas ajustadas o el método de Bucher7,8—, la única manera fiable de establecer diferencias de eficacia entre los fármacos debería ser mediante comparaciones directas. En este sentido, si bien se ha publicado alguna comparación indirecta entre estudios9, las diferencias mencionadas en cuanto al tipo de población, el control y el diseño del estudio propician que tales comparaciones estén sometidas a ciertos sesgos, por lo que hay que interpretarlas con precaución.
CONCLUSIONESLa aparición de los nuevos anticoagulantes orales como tratamiento de prevención de fenómenos de tromboembolia en la fibrilación auricular no valvular ha supuesto sin duda un significativo avance. Se ha estudiado la eficacia y la seguridad de los tres nuevos fármacos en ECA de no inferioridad (hipótesis principal), en los que también se analizaban hipótesis de superioridad. Aunque existe un lógico entusiasmo por conocer qué fármaco es más eficaz, efectivo y eficiente, sin las pertinentes comparaciones directas no se podrá obtener de manera fiable esa información. En todo caso, resulta razonable pensar que, conforme vaya pasando el tiempo y se tenga más datos de estudios observacionales, se irán definiendo las características de la enfermedad —pero, sobre todo, del paciente (comorbilidades, riesgo hemorrágico, factores psicosociales, etc.)— y del entorno que definirán las indicaciones precisas de cada fármaco para grupos de pacientes concretos.
CONFLICTO DE INTERESESEl autor ha percibido honorarios por cursos docentes y por charlas académicas de Boehringer-Ingelheim, Bayer y Pfizer.
Full English text available from: www.revespcardiol.org/en
El autor agradece a Content Ed Net por su asistencia editorial para editar el manuscrito.