
El solapamiento fenotípico entre cardiopatía congénita, miocardiopatía no compactada y miocardiopatía arritmogénica es infrecuente. Bermúdez-Jiménez et al.1 publicaron en Revista Española de Cardiología un artículo en el que se describe a una familia portadora de una mutación en NKX2.5 (p.Glu167Lys) que muestra este fenotipo. Consideramos oportuno incidir en el riesgo de muerte súbita asociado a mutaciones en NKX2.5 asociadas a este fenotipo, para lo que describimos a una familia en seguimiento en nuestro centro.
El gen NKX2.5 codifica un factor de transcripción implicado en el desarrollo cardiaco que contiene 3 dominios2. El homeodominio es necesario para la interacción con el ADN y otros factores de transcripción. Las mutaciones en NKX2.5 se han asociado a defectos cardiacos septales, defectos de conducción y miocardiopatía no compactada.
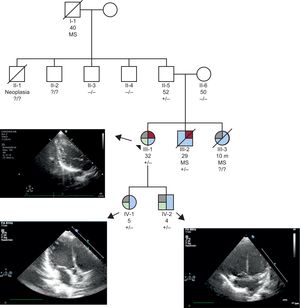
Se presenta el caso de una mujer de 30 años, cuyo abuelo paterno falleció súbitamente durante el sueño a los 40 años, enviada para estudio después de que su hermano sufriera una parada cardiaca. Su hermana falleció súbitamente en edad pediátrica en el posoperatorio de reparación de una comunicación interauricular (CIA). Su hermano sufrió una parada cardiaca durante el sueño, con asistolia como primer ritmo observado y fibrilación ventricular tras las maniobras de reanimación. Se detectó en el ecocardiograma leve dilatación de ventrículo izquierdo, con criterios de ausencia de compactación, disfunción sistólica moderada-grave y aneurisma del septo interauricular sin shunt. Se describe electrocardiograma normal. El paciente falleció por encefalopatía hipóxico-isquémica. No se realizó autopsia. El padre y la madre de la paciente no mostraron alteraciones en el ecocardiograma ni en el Holter de 24 h.
Se reparó a la paciente una CIA ostium secundum en la infancia mediante cirugía. Se realizó una cardiorresonancia, que mostró ausencia de compactación del ventrículo izquierdo con fracción de eyección en el límite bajo de la normalidad. Se apreciaba bloqueo auriculoventricular de primer grado, sin otras alteraciones en el Holter.
La paciente es madre de 2 hijos (5 y 4 años) diagnosticados de CIA tipo ostium secundum con hipertrabeculación en ecocardiograma, sin criterios de no compactación y PR alargado para su edad, no intervenidos hasta el momento del defecto.
Se realizó estudio genético al hermano fallecido mediante un panel de secuenciación de nueva generación; se incluyeron los exones de 268 genes y se centró el análisis en 16 (ACTC1, CASQ2, DMD, DTNA, HCN4, LDB3, LMNA, MYBPC3, MYH7, NKX2-5, SCN5A, TAZ, TNNI3, TNNT2, TPM1 y VCL). Se identificó una variante en heterocigosis en NKX2.5 (p.Lys183Asn), no descrita en bases de datos públicas de población general ni indicada previamente y deletérea según predictores bioinformáticos. La variante p.Lys183Asn afecta a un residuo altamente conservado evolutivamente (figura 1). Resultaron portadores de la mutación nuestra paciente, sus hijos y su padre. La madre no es portadora. Se realizó cardiorresonancia al padre, sin hallazgos relevantes. Se estudió también a 2 tíos, que resultaron negativos, y se demostró como único hallazgo positivo (ECG, imagen) hipertrabeculación apical en 1 de ellos (figura 2).


Pedigrí de la familia. Granate: miocardiopatía no compactada; gris: CIA; verde: bloqueo auriculoventricular. Los heterocigotos se marcan como ±. Imágenes ecocardiográficas de 3 de los probandos. CIA: comunicación interauricular; MS: muerte súbita. Esta figura se muestra a todo color solo en la versión electrónica del artículo.
La evidencia de cosegregación de la mutación con el fenotipo en 4 sujetos afectados de la familia, con expresividad y penetrancia variables, provee evidencia sobre su probable patogenicidad. El hallazgo de un portador carente de fenotipo es compatible con la penetrancia incompleta demostrable en otras mutaciones en NKX2.5. Es pertinente incluir en el diagnóstico diferencial de la muerte súbita del sujeto I-1 otras causas como la cardiopatía isquémica.
p.Lys183Asn se localiza en el homeodominio de NKX2.5 (aminoácidos 138-197). Unas pocas variantes en esta región se han asociado al desarrollo de defectos del septo interauricular y defectos de la conducción, con penetrancia y expresividad variables, la mayoría de ellas con evidencia de cosegregación (i.e. p.Leu171Pro3, p.Arg190His3).
La única mutación en NKX2.5 asociada hasta la descripción de p.Glu167Lys1 con un fenotipo compuesto de no compactación, CIA y defectos de la conducción y muerte súbita se localiza en este dominio de la proteína (p.Leu171Argfs*7). Esta variante fue descrita por Ouyang et al.4 en una familia americana con penetrancia y expresividad variables, con un portador que falleció súbitamente.
Maury et al.5 presentaron en el congreso de la Sociedad Europea de Cardiología una serie de 48 pacientes (18 familias) portadores de mutaciones en NKX2.5, en la que describieron 8 muertes súbitas. El 75% de los portadores tenían CIA y el 90%, defectos de conducción5. Cinco de los portadores cumplían criterios de miocardiopatía no compactada.
En nuestra paciente se implantó un desfibrilador monocameral como prevención primaria, en el que se registraron episodios recurrentes no sostenidos de taquicardia ventricular polimorfa y 2 descargas apropiadas del dispositivo tras degeneración a fibrilación ventricular. Se inició tratamiento con sotalol y no ha habido recurrencias en los últimos 7 meses.
Esta familia, junto con la de Bermúdez-Jiménez et al.1, propone que las mutaciones en esta región de NKX2.5 (homeodominio) conllevan defectos del septo interauricular y el sistema de conducción y ausencia de compactación ventricular, junto con una elevada carga de muerte súbita. A diferencia de esta familia con hallazgos compatibles con miocardiopatía arritmogénica (portadores de mutación en DSP), no existe en esta un segundo evento mutacional relacionable con riesgo de muerte súbita, que haría más plausible esta asociación.
La detección de ausencia de compactación en pacientes con CIA podría poner sobre aviso de la posible presencia de esta entidad, de comportamiento maligno.