Avances recientes en genética han permitido el descubrimiento de nuevos genes relacionados con la hipertensión arterial pulmonar, como TBX4 y KCNK3. El fenotipo y el pronóstico asociado a ellos se han detallado escasamente y se desconoce su papel en la población española. El objetivo de este estudio es caracterizar genotípicamente una cohorte española de pacientes con hipertensión arterial pulmonar idiopática y hereditaria, describiendo el fenotipo y los factores pronósticos asociados a BMPR2 y a los nuevos genes (KCNK3 y TBX4).
MétodosSe seleccionó a 165 pacientes adultos con hipertensión arterial pulmonar: 143 con hipertensión arterial pulmonar idiopática y 22 con hipertensión arterial pulmonar familiar. Se compararon las características basales y la supervivencia libre de eventos entre los distintos subgrupos, se analizaron los factores predictores de mal pronóstico y se llevó a cabo el cribado familiar.
ResultadosEl estudio genético fue positivo en 16 pacientes con hipertensión arterial pulmonar idiopática (11,10%) y 15 con hipertensión arterial pulmonar familiar (68,18%), y se hallaron 19 mutaciones en BMPR2, 4 en TBX4 y 3 en KCNK3. Se observó mayor supervivencia libre de eventos en las formas asociadas a TBX4 (p < 0,01). El diagnóstico en clases funcionales avanzadas fue el único factor pronóstico en las formas heredables. El cribado de familiares fue positivo en el 37,5%.
ConclusionesEn la población española con hipertensión arterial pulmonar puede existir un sustrato genético diferente, con menor proporción de mutaciones en BMPR2. A la vista de nuestros resultados, las formas asociadas a TBX4 podrían conllevar un fenotipo más benigno, y el diagnóstico tardío sería un factor de mal pronóstico en las formas heredables de la enfermedad.
Palabras clave
La hipertensión arterial pulmonar (HAP) es una enfermedad infrecuente de pronóstico ominoso en ausencia de tratamiento. La HAP puede presentarse en diferentes formas, entre ellas la HAP idiopática (HAPI) en ausencia de causa conocida y la HAP hereditaria (HAPH) en caso de relacionarse con una alteración genética o de que haya agregación familiar.
La HAPI es una enfermedad rara que afecta a 5,6 adultos/millón de habitantes en España, con 1,2 casos nuevos por millón de habitantes al año, según datos del Registro Español de Hipertensión Arterial Pulmonar (REHAP), registro voluntario español de pacientes con HAP en marcha desde el año 2007, cuya metodología se ha descrito ampliamente1.
El primer gen relacionado con el desarrollo de HAPH fue BMPR2, que codifica el receptor de la proteína morfogenética tipo 2 y regula múltiples funciones celulares2. Se han descrito mutaciones en BMPR2 en el 75% de las formas familiares y el 25% de las idiopáticas, que muestran un patrón de herencia autosómico dominante con penetrancia incompleta (∼20%) variable según el sexo (el 42% de las mujeres frente al 14% de los varones)2,3 y expresividad variable4–7, y se desconocen los mecanismos moleculares subyacentes8–12.
Se conocen más de 300 mutaciones en BMPR2, a lo largo de sus 13 exones y 4 dominios proteicos4,5, la mayoría de ellas (70%) tipo frame-shift, nonsense o deleciones4 y una minoría, tipo missense (30%).
Avances recientes en genética, como la secuenciación masiva, han permitido el descubrimiento de nuevos genes relacionados con la HAP4,13–20, tales como KCNK3, que codifica un canal de potasio dependiente de pH13, TBX4, que codifica el factor de transcripción TBX414 implicado en el desarrollo embrionario y relacionado con el síndrome de small patella, o los genes CAV115, TOPBP1, SMADS o NOTCH34,13–20, entre otros. Además, se ha relacionado EIF2AK4 con el desarrollo de formas recesivas de enfermedad venooclusiva pulmonar16,17. La HAP se ha convertido, por lo tanto, en una enfermedad genéticamente compleja con un creciente número de genes implicados, y escasea el conocimiento acerca del fenotipo asociado a cada uno de ellos.
Los objetivos de este trabajo son caracterizar genética y fenotípicamente una cohorte española de pacientes con HAPI y HAPH, analizar el impacto pronóstico de las diversas alteraciones genéticas, los factores predictores de muerte o entrada en lista de espera de trasplante pulmonar en esta población y el impacto familiar en términos de penetrancia y dinámica del cribado de familiares.
MÉTODOSEl estudio multicéntrico español de genética de HAP se inició en noviembre de 2011. Se trata de un estudio observacional y ambispectivo de pacientes adultos con HAPI/HAPH atendidos en 2 centros españoles (Hospital Universitario 12 de Octubre y Hospital Universitario Vall d’Hebron, e incluidos en el Biobanc del Hospital Clínic de Barcelona). Para el presente análisis se excluyó a los pacientes con enfermedad venooclusiva pulmonar, y se conformaron 2 cohortes: una de pacientes consecutivos atendidos en el Hospital Universitario 12 de Octubre y el Hospital Universitario Vall d’Hebron (enero de 2011 a mayo de 2015) y otra de pacientes incluidos en el Biobanc del Hospital Clínic de Barcelona (enero de 2013 a marzo de 2014). El diagnóstico de HAP se realizó según recomendaciones internacionales21. Según hubiera historia familiar de HAP, previamente al análisis genético se clasificó a los pacientes en HAP familiar (historia familiar positiva) o HAPI (historia familiar negativa), y tras el análisis genético se los reclasificó en formas idiopáticas (historia familiar negativa y sin alteraciones genéticas) y heredables (pacientes con historia familia positiva y análisis genético positivo, con historia familiar positiva y análisis genético negativo y pacientes con historia familiar negativa pero análisis genético positivo). Los datos clínicos se obtuvieron del REHAP y el análisis genético se llevó a cabo en el Instituto de Genética Médica y Molecular (INGEMM) del Hospital Universitario La Paz.
Caracterización clínicaSe analizaron datos demográficos (sexo y edad), clínicos (clase funcional de la New York Heart Association [NYHA], prueba de marcha de 6 minutos, síncope), hemodinámicos (presión en aurícula derecha, presión arterial pulmonar media, índice cardiaco y resistencia vascular pulmonar arteriolar, respuesta al test agudo vasodilatador) y parámetros de función pulmonar (flujo espiratorio en 1 s, capacidad vital forzada y capacidad de difusión de monóxido de carbono de todos los pacientes al diagnóstico. Se realizó cribado clínico y radiológico del síndrome de small patella en portadores de mutaciones en TBX4.
Estudio molecularSe llevó a cabo la secuenciación de los exones y las uniones intrón-exón de BMPR2, TBX4 y KCNK3 y el estudio de reordenamientos cromosómicos mediante multiplex ligation-dependent probe amplification (MLPA) MLPA (multiplex ligation-dependent probe amplification) (anexo 1 del material suplementario).
Las variantes identificadas se clasificaron según su frecuencia en población general considerando mutaciones a las ausentes o con frecuencias alélicas muy bajas (< 0,01%) en bases de datos públicas (Exome Variant Server, 1000G y ExAC1S-3S del material suplementario). Se evaluó la patogenicidad de las variantes teniendo en cuenta, además, su efecto en la proteína, su publicación previa en la bibliografía, la evidencia de cosegregación con la enfermedad y los resultados de predictores bioinformáticos (Polyphen-2, MutationTaster4S,5S delmaterial suplementario, SIFT [Sorting Intolerant from Tolerant], MutPred, SNPs&GO). Así, las variantes encontradas se clasificaron en patogénicas, posiblemente patogénicas, variantes de significado incierto y variantes no patogénicas (anexo 2 del material suplementario). Una vez completado el estudio de los probandos, se llevó a cabo el estudio genético de familiares de primer grado de pacientes portadores de mutación y se elaboró el árbol genealógico.
Análisis estadísticoSe utilizaron los programas Stata (versión 13, Stata Corp.; College Station, Texas, Estados Unidos) e IBM SPSS 22 (SPSS, Inc.; Chicago, Illinois, Estados Unidos), y los datos se presentaron como media ± desviación estándar con nivel de significación en p ≤ 0,05. Se realizó un análisis de supervivencia univariante de Kaplan-Meier tomando como origen del seguimiento el momento del diagnóstico y el final del seguimiento el día 15 de junio de 2015, con un seguimiento máximo de 15 años (censura tipo I generalizada). Se utilizó el log rank test para la comparación de supervivencias de los pacientes con HAPI y HAPH y las formas asociadas a los distintos genes; el valor del evento se definió como muerte o entrada en la lista de espera de trasplante pulmonar. Se calculó la hazard ratio y el intervalo de confianza del 95% de los factores clásicos (presión en aurícula derecha, índice cardiaco, prueba de marcha de 6 minutos, clase funcional de la NYHA al diagnóstico, sexo, presión pulmonar media arterial pulmonar y resistencia vascular pulmonar) y se incluyó la etiología heredable. Además, se evaluó la interacción entre los factores clásicos y el hecho de ser heredable. Las variables que en el análisis univariable mostraron un nivel de significación de p < 0,10 se incluyeron en el modelo de regresión de Cox multivariable, se aplicaron un método de selección por pasos hacia delante y un método de selección por pasos hacia atrás para confirmar los resultados.
El estudio se ajusta a los principios de la Declaración de Helsinki y a la ley de protección de datos personales y fue aprobado por los comités éticos de los centros participantes. Todos los pacientes otorgaron su consentimiento informado.
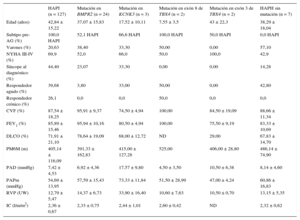
RESULTADOSSe incluyó a 165 pacientes con diagnóstico de HAP entre el 1 de noviembre de 2011 y el 1 de mayo de 2015; 143 (86,6%) sufrían HAPI y 22 (13,3%), HAP familiar. En la tabla 1 se muestran las características basales.
Características basales de la población, hipertensión arterial pulmonar heredable frente a hipertensión arterial pulmonar idiopática
| Población total (n = 165) | HAPH (n = 38) | HAPI (n = 127) | p | |
|---|---|---|---|---|
| Subtipo pre-AG | 143 HAPI, 22 HAPH | 16 HAPI, 22 HAPH | 127 HAPI | — |
| Edad (años) | 40,68 ± 16,13 | 35,18 ± 17,51 | 42,84 ± 15,22 | 0,006 |
| Varones (%) | 24,8 | 38,4 | 20,63 | 0,02 |
| NYHA (%) | 0,054 | |||
| I-II | 33,4 | 43,6 | 30,1 | |
| III | 61,8 | 43,6 | 66,6 | |
| IV | 4,2 | 8,1 | 3,1 | |
| Síncope al diagnóstico (%) | 38,2 | 17,9 | 44,4 | 0,003 |
| Respondedor al test agudo (%) | 33,30 | 12,82 | 39,68 | 0,001 |
| Respondedor crónico (%) | 20,60 | 2,63 | 26,10 | 0,005 |
| CVF (%) | 88,89 ± 16,9 | 93,67 ± 11,35 | 87,54 ± 18,25 | 0,03 |
| FEV1(%) | 87,29 ± 14,89 | 92,44 ± 12,1 | 85,89 ± 15,46 | 0,01 |
| DLCO (%) | 72,23 ± 20,67 | 76,00 ± 24,14 | 71,91 ± 21,10 | NS |
| PM6M (m) | 410,61 ± 116,76 | 417,19 ± 106,64 | 405,14 ± 116,09 | NS |
| PAD (mmHg) | 7,59 ± 4,79 | 8,16 ± 5,58 | 7,42 ± 4,53 | NS |
| PAPm (mmHg) | 55,77 ± 14,43 | 59,28 ± 15,54 | 54,69 ± 13,95 | NS |
| IC (l/m/m2) | 2,34 ± 0,72 | 2,31 ± 0,67 | 2,36 ± 0,67 | NS |
| RVP (UW) | 13,67 ± 10,93 | 15,57 ± 9,03 | 12,79 ± 5,47 | 0,02 |
CVF: capacidad vital forzada; DLCO: capacidad de difusión de monóxido de carbono; FEV1: volumen espiratorio forzado en el primer segundo; HAPH: hipertensión arterial pulmonar heredable; HAPI: hipertensión arterial pulmonar idiopática; IC: índice cardiaco; NS: no significativo; NYHA: New York Heart Association; PAD: presión auricular derecha; PAPm: presión arterial pulmonar media; PM6M: prueba de marcha de 6 minutos; RVP: resistencia vascular pulmonar arteriolar; Subtipo pre-AG: subtipo de HAP previo al análisis genético.
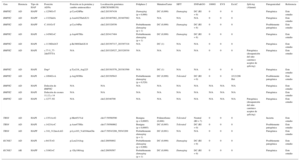
El estudio genético fue positivo para alguna variante en los tres genes analizados en 31 probandos (18,78%): 16 con HAPI (11,10%) y 15 con HAP familiar (68,18%). Se identificaron 19 variantes en BMPR2 (tabla 1S del material suplementario) en 24 probandos, 3 variantes en TBX4 en 4 probandos y 2 variantes en KCNK3 en 3 probandos (tabla 2S del material suplementario). En la tabla 2 se muestran las características basales según el gen afectado.
Características basales según el gen afectado
| HAPI (n = 127) | Mutación en BMPR2 (n = 24) | Mutación en KCNK3 (n = 3) | Mutación en exón 8 de TBX4 (n = 2) | Mutación en exón 3 de TBX4 (n = 2) | HAPH sin mutación (n = 7) | |
|---|---|---|---|---|---|---|
| Edad (años) | 42,84 ± 15,22 | 37,07 ± 15,83 | 17,52 ± 10,11 | 7,55 ± 3,5 | 43 ± 22,3 | 38,29 ± 18,04 |
| Subtipo pre-AG (%) | 100,0 HAPI | 52,1 HAPI | 66,6 HAPI | 100,0 HAPI | 50,0 HAPI | 0,0 HAPI |
| Varones (%) | 20,63 | 38,40 | 33,30 | 50,00 | 0,00 | 57,10 |
| NYHA III-IV (%) | 69,9 | 52,0 | 66,0 | 50,0 | 100,0 | 42,9 |
| Síncope al diagnóstico (%) | 44,40 | 23,07 | 33,30 | 0,00 | 0,00 | 14,28 |
| Respondedor agudo (%) | 39,68 | 3,80 | 33,00 | 50,00 | 0,00 | 42,80 |
| Respondedor crónico (%) | 26,1 | 0,0 | 0,0 | 50,0 | 0,0 | 0,0 |
| CVF (%) | 87,54 ± 18,25 | 95,91 ± 9,37 | 74,50 ± 4,94 | 100,00 | 84,50 ± 19,09 | 88,66 ± 11,34 |
| FEV1 (%) | 85,89 ± 15,46 | 95,94 ± 10,16 | 80,50 ± 4,94 | 100,00 | 75,50 ± 9,19 | 83,33 ± 10,69 |
| DLCO (%) | 71,91 ± 21,10 | 78,64 ± 19,09 | 68,00 ± 12,72 | ND | 29,00 | 67,83 ± 14,70 |
| PM6M (m) | 405,14 ± 116,09 | 391,33 ± 162,83 | 415,00 ± 127,28 | 525,00 | 406,00 ± 28,80 | 486,14 ± 74,90 |
| PAD (mmHg) | 7,42 ± 4,53 | 6,92 ± 4,36 | 17,57 ± 9,80 | 4,50 ± 3,50 | 10,50 ± 6,38 | 8,14 ± 4,60 |
| PAPm (mmHg) | 54,69 ± 13,95 | 57,59 ± 15,43 | 73,33 ± 11,84 | 51,50 ± 28,99 | 47,00 ± 4,24 | 60,86 ± 16,83 |
| RVP (UW) | 12,79 ± 5,47 | 14,37 ± 6,73 | 33,90 ± 16,40 | 10,60 ± 7,63 | 10,50 ± 0,70 | 13,15 ± 5,35 |
| IC (l/m/m2) | 2,36 ± 0,67 | 2,33 ± 0,75 | 2,44 ± 1,01 | 2,60 ± 0,42 | ND | 2,32 ± 0,62 |
CVF: capacidad vital forzada; DLCO: capacidad de difusión de monóxido de carbono; FEV1: volumen espiratorio forzado en el primer segundo; HAPF: hipertensión arterial pulmonar heredable; HAPI: hipertensión arterial pulmonar idiopática; IC: índice cardiaco; ND: no disponible; NYHA: New York Heart Association; PAD: presión auricular derecha; PAPm: presión arterial pulmonar media; PM6M: prueba de marcha de 6 minutos; RVP: resistencia vascular pulmonar arteriolar; Subtipo pre-AG: subtipo de HAP previo al análisis genético.
El estudio genético fue positivo para BMPR2 en el 9,1% de los pacientes con HAPI y el 50,0% de los pacientes con HAPH. Se encontraron 19 variantes en BMPR2 (8 de ellas, de tipo missense) en 24 probandos pertenecientes a 20 familias no relacionadas; 2 probandos no relacionados compartían la misma variante (p.Asp491Glu) (tabla 1S del material suplementario). Ocho de las variantes encontradas ya estaban descritas en la literatura, frente a 11 no descritas, 9 de estas consideradas patogénicas por tratarse de 7 cambios radicales (frameshift, variantes estructurales o variantes intrónicas con probable efecto en el splicing) y 2 variantes descritas localizadas en residuos con mutaciones en pacientes con HAP (p.Cys420Phe, localizada en el mismo codón que p.Cys420Tyr12S,13S del material suplementario y p.Cys420Arg14S,15S del material suplementario, y p.As487His, localizada en el mismo codón que p.Asp487Val9S del material suplementario,). A las 2 restantes (p.Cys34Phe y p.Arg365His) se las consideró posiblemente patogénicas, ya que los predictores bioinformáticos indican un efecto deletéreo. Solo se pudo demostrar cosegregación en una familia (p.Asn442ThrfsX31). Para el resto de las variantes, no se descartó cosegregación, pero no se pudo confirmar por la penetrancia incompleta asociada a las mutaciones en BMPR2 (tabla 3). La tabla 3S del material suplementario recoge las características basales según el tipo de mutación.
Resultados del estudio de patogenicidad mediante herramientas bioinformáticas de aquellas variantes que no han sido descritas previamente
| Gen | Herencia | Tipo de HAP | Posición ADNc | Posición en la proteína y cambio aminoacídico | Localización genómica (GRCH38/HG38) | Poliphen-2 | MutationTaster | SIFT | SNPs&GO | 1000G | EVS | ExAC | Splicing (Alamut) | Patogenicidad | Referencia |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BMPR2 | AD | HAPF | c.1259G>T | p.Cys420Phe | chr2:203397438 | Damaging (p = 0,990) | DC (0,999) | Damaging | DC (RI = 9) | 0 | 0 | 0 | Patogénica | Este estudio | |
| BMPR2 | AD | HAPF | c.1325delA | p.Asn442ThrfsX31 | chr2:203407082_203407082 | N/A | N/A | N/A | N/A | 0 | 0 | 0 | Patogénica | Este estudio | |
| BMPR2 | AD | HAPF | C.101G>T | p.Cys34Phe | chr2:203329556 | Probablemente damaging (p = 1) | DC (0,999) | Damaging | DC (RI = 8) | 0 | 0 | 0 | Posiblemente patogénica | Este estudio | |
| BMPR2 | AD | HAPI | c.1459G>C | p.Asp487His | chr2:203417484 | Probablemente damaging (p = 1) | DC (0,999) | Damaging | DC (RI = 8) | 0 | 0 | 0 | Patogénica | Este estudio | |
| BMPR2 | AD | HAPI | c.1138DelAT | p.Ile380GlnfsX18 | chr2:203397317_203397318 | N/A | DC (1) | N/A | N/A | 0 | 0 | 0 | Patogénica | Este estudio | |
| BMPR2 | AD | HAPI | c.77-5_77-2delTTTA | N/A | chr2:203329527_203329530 | N/A | N/A | N/A | N/A | 0 | 0 | 0 | Patogénica (desaparición del sitio canónico aceptor de splicing) | Patogénica | Este estudio |
| BMPR2 | AD | HAPI | Dup* | p.Tyr218_Arg225 | chr2:203383576_203383599 | N/A | DC (1) | N/A | N/A | 0 | 0 | 0 | Patogénica | Este estudio | |
| BMPR2 | AD | HAPI | c.1094G>A | p.Arg365His | chr2:203395643 | Probablemente damaging (p = 0,529) | DC (0,999) | Tolerated | DC (RI = 0) | 0 | 0 | 2/121296 Het | Posiblemente patogénica | Este estudio | |
| BMPR2 | AD | HAPI | Deleción de BMPR2 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | Patogénica | Este estudio | |
| BMPR2 | AD | HAPI | Deleción de exones 11,12 y 14 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | Patogénica | Este estudio | |
| BMPR2 | AD | HAPI | c.1277-3G | N/A | chr2:20340700 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | Patogénica (desaparición del sitio canónico aceptor de splicing) | Patogénica | Este estudio |
| TBX4 | AD | HAPI | c.1351A>G | p.Met451Val | chr17:59560590 | Benigna (p < 0,0005) | Polimorfismo (0,888) | Tolerated | Neutral (RI = 7) | 0 | 0 | 0 | Incierta | Este estudio | |
| TBX4 | AD | HAPI | c.1423A>C | p.Asn475His | chr17:59560662 | Benigna (p < 0,0005) | DC (0,995) | Tolerated | DC (RI = 0) | 0 | 0 | 0 | Posiblemente patogénica | Este estudio | |
| TBX4 | AD | HAPF | c.310_312insAAG | p.Lys103_Val104insGlu | chr17:59543208_59543209 | Probablemente damaging (p = 1) | DC (0,991) | N/A | N/A | 0 | 0 | 0 | Patogénica | Este estudio | |
| KCNK3 | AD | HAPI | c.641T>G | p.Leu214Arg | chr2:26950892 | Probablemente damaging (p = 0,998) | DC (0,999) | Damaging | DC (RI = 9) | 0 | 0 | 0 | Posiblemente patogénica | Este estudio | |
| KCNK3 | AR | HAPF | c.316G>C | p. Gly106Arg | chr2:26950567 | Probablemente damaging (p = 1) | DC (0,999) | Damaging | DC (RI = 9) | 0 | 0 | 0 | Patogénica | Este estudio |
AD: herencia autosómica dominante; AR: herencia autosómica recesiva; DC: disease causing; Dup*: duplicación c.653_676dupATAAAGGCTCCTTGGATGAGCG; HAPF: hipertensión arterial pulmonar familiar; HAPI: hipertensión arterial pulmonar idiopática; N/A: no aplicable; RI: índice de confianza; SIFT: Sorting Intolerant from Tolerant.
Se han aplicado diversos predictores de patogenicidad cuyos ratios son: Polyphen-2 (tiene una escala de 0 a 1: aquellos valores cercanos a 1 tienen mayor probabilidad de ser clasificados como patogénicos [«damaging» en la tabla]); MutationTaster (su escala también va de 0 a 1 y aquellos valores cercanos a 1 se correlacionan con una mayor posibilidad de patogenicidad); SIFT (se ha clasificado las variantes como «damaging» si es posiblemente patogénica y «tolerated» si es probable que la variante sea benigna); SNPs&GO (calcula la posibilidad de patogenicidad con un ratio de confianza de la predicción que va de 1 a 9, siendo 1 el valor más bajo de confianza y 9 el valor máximo de confianza). Además, se han analizado estas variantes en varias poblaciones control como aparece en la tabla: EVS (Exome Variant Server), 1000G (1000 Genomes database) y ExAC (Exome Agregation Consortium). Valores iguales a 0 significa que el cambio concreto no está presente en esa base de datos, para aquellos que sí aparecen se muestra el conteo alélico respecto al total.
Transcrito utilizado para analizar TBX4: ENST00000240335. Transcrito utilizado para analizar BMPR2: ENST00000374580. Transcrito utilizado para analizar KCNK3: ENST00000302909.
Se identificaron 2 variantes de tipo missense (p.Leu214Arg y p.Gly106Arg) en el exón 2 de KCNK3, no descritas en la literatura, en 1 paciente con HAPI y 2 pacientes con HAPH relacionados entre sí. El cambio p.Gly106Arg se identificó en homocigosis en un probando con una forma agresiva y se lo consideró patogénico, dada su cosegregación con la enfermedad. La mutación p.Leu214Arg se clasificó como posiblemente patogénica, y su análisis in silico indica un efecto probablemente patogénico (tabla 3 y tabla 2S del material suplementario).
Mutaciones en TBX4Se detectaron 2 variantes de tipo missense (p.Met451Val y p.Asn475His) en el exón 8 de TBX4 en 2 pacientes con HAPI y una inserción de tres nucleótidos en el exón 3 en 2 pacientes con HAPH relacionados entre sí. Esta variante se consideró patogénica. La variante p.Asn475His se consideró posiblemente patogénica porque el análisis bioinformático indicó un efecto deletéreo. La variante p.Met451Val se consideró de significado incierto y se la clasificó como benigna por los predictores de patogenicidad, y aunque no aparece en las poblaciones de control analizadas, el aminoácido no está muy conservado a lo largo de la evolución. Todo esto parece indicar que esta variante probablemente sea benigna, ya que por sí sola no parece que comprometa la función proteica y la posterior aparición del fenotipo asociado. Ninguno de los pacientes reunía criterios de síndrome de small patella (tabla 3 y tabla 2S del material suplementario).
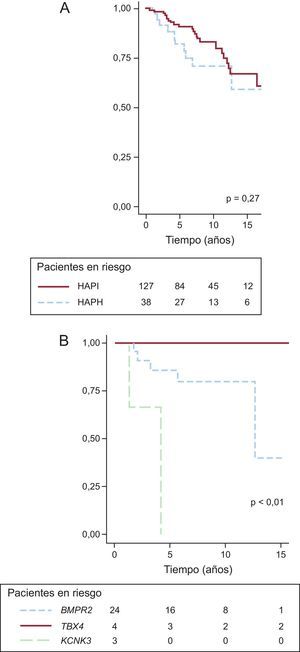
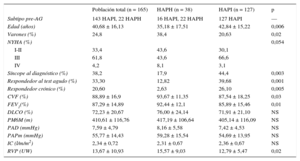
Impacto pronóstico de las alteraciones genéticas (análisis de supervivencia Kaplan-Meier)La media de seguimiento fue 7,74 ± 4,46 años. El análisis de supervivencia no mostró diferencias significativas entre HAPI y HAPH (figura A), y hubo diferencias significativas solo entre las formas de HAPH asociadas a TBX4, KCNK3 y BMPR2 (figura B).
Análisis de factores predictores de muerte o trasplante (regresión de Cox)
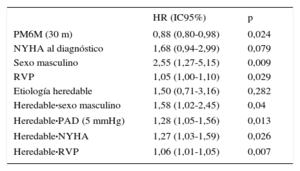
En la tabla 4 y en la tabla 5 se muestran los resultados de los análisis univariable y multivariable (log función de verosimilitud = –103,29; log rank χ2 = 13,03; p = 0,005).
Análisis univariable de Cox
| HR (IC95%) | p | |
|---|---|---|
| PM6M (30 m) | 0,88 (0,80-0,98) | 0,024 |
| NYHA al diagnóstico | 1,68 (0,94-2,99) | 0,079 |
| Sexo masculino | 2,55 (1,27-5,15) | 0,009 |
| RVP | 1,05 (1,00-1,10) | 0,029 |
| Etiología heredable | 1,50 (0,71-3,16) | 0,282 |
| Heredable·sexo masculino | 1,58 (1,02-2,45) | 0,04 |
| Heredable·PAD (5 mmHg) | 1,28 (1,05-1,56) | 0,013 |
| Heredable·NYHA | 1,27 (1,03-1,59) | 0,026 |
| Heredable·RVP | 1,06 (1,01-1,05) | 0,007 |
IC95%: intervalo de confianza del 95%; HR: hazard ratio; NYHA: New York Heart Association; PAD: presión auricular derecha; PM6M: prueba de marcha de 6 minutos; RVP: resistencia vascular pulmonar arteriolar.
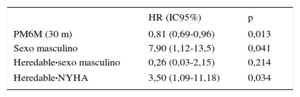
Análisis multivariable de Cox
| HR (IC95%) | p | |
|---|---|---|
| PM6M (30 m) | 0,81 (0,69-0,96) | 0,013 |
| Sexo masculino | 7,90 (1,12-13,5) | 0,041 |
| Heredable·sexo masculino | 0,26 (0,03-2,15) | 0,214 |
| Heredable·NYHA | 3,50 (1,09-11,18) | 0,034 |
HR: hazard ratio; IC95%: intervalo de confianza del 95%; NYHA: New York Heart Association; PM6M: prueba de marcha de 6 minutos.
Rechazaron el estudio genético 2 de las 19 familias a las que se lo propuso, y les aceptaron 72 de los 90 familiares (80%) de las 17 familias restantes. Se encontró a 21 portadores sanos (29,1%) y 6 portadores afectados (8,3%). Las características de los portadores se muestran en las tablas 4S y 5S del material suplementario. La penetrancia estimada de las mutaciones en BMPR2 fue del 23,5%; en KCNK3, del 25,0%, y en TBX4, del 33,0%.
DISCUSIÓNLa nuestra, con 165 pacientes, es la más amplia serie de pacientes con HAP estudiados genéticamente en España, y recoge el 33% de las formas idiopáticas y heredables registradas en el REHAP hasta noviembre de 20141 y el 49% de los pacientes atendidos en 3 centros de referencia. Hemos hallado mutaciones en BMPR2 en el 50% de las formas familiares y el 9% de las formas idiopáticas, cifras inferiores al 75 y el 25% publicados previamente2,4,5,7,12. Si se incluyen las mutaciones en TBX4 y KCNK3, los pacientes portadores de alguna mutación aumentan al 68,1 y el 11,1% respectivamente, lo que se acerca a los datos publicados por otros grupos12. Esto podría traducir la existencia de un sustrato genético distinto en la población española, en la que el porcentaje total de pacientes portadores de mutaciones es similar que en otras poblaciones, pero con una distribución de los genes afectados distinta4,7,19,22. No obstante, son necesarios estudios más extensos para confirmar esta hipótesis.
Nuestro trabajo describe el fenotipo asociado a cada tipo de alteración de BMPR2, así como de las formas asociadas a KCNK3 y TBX4, mucho menos conocidas. El análisis de nuestros datos revela un perfil más agresivo de las formas asociadas a KCNK3 frente a un comportamiento más benigno de TBX4, con un perfil clínico intermedio de BMPR2. Sin embargo, de manera análoga a publicaciones previas12, no se ha encontrado diferencias en la supervivencia entre las formas heredables y las idiopáticas. Uno de los hallazgos más relevantes de este estudio es el marcado impacto pronóstico en las formas heredables del diagnóstico en clases funcionales avanzadas, lo cual pone de manifiesto la importancia de lograr un diagnóstico precoz especialmente en este subgrupo de pacientes.
Hipertensión arterial pulmonar familiar frente a idiopáticaEn nuestra serie, el perfil clínico de las formas heredables en su conjunto es similar al asociado a BMPR2 descrito previamente4,7,9,12,19. Destaca que el 31,81% de los pacientes con HAPH presentan un estudio genético negativo, datos superiores a lo publicado2,3.
Mutaciones en BMPR2Se ha encontrado 19 mutaciones en BMPR2, con un 42% de mutaciones de tipo missense, cifras algo superiores a lo descrito4. Once de ellas no han sido descritas previamente, lo cual pone de manifiesto la alta complejidad de este gen, del que continúan apareciendo nuevas alteraciones5,20 a pesar de haberse estudiado ampliamente. Si bien estos cambios se distribuyen por todo el gen afectando a los distintos dominios proteicos, 8 de ellos (42%), una frecuencia mucho mayor que la descrita4, se localizan en el exón 2, lo cual podría traducir la existencia de un «punto caliente» para mutaciones en dicho exón, al menos en la población española.
Cabe destacar el hallazgo de la mutación c.653_676dupATAAAGGCTCCTTGGATGAGCG, una duplicación de 22 pares de bases, ya que nunca antes se había descrito este tipo de cambio en relación con el desarrollo de HAP. Esta mutación podría caracterizarse por una mayor penetrancia (el 50,0 frente al 23,5%), un hallazgo novedoso con posibles implicaciones futuras.
En cuanto al fenotipo asociado a BMPR2, los hallazgos descritos hasta la fecha se confirman en nuestra serie (tabla 2)4–9,20, y llama la atención la menor proporción de mujeres en las formas hereditarias de la enfermedad, hecho nunca antes indicado. Se confirma la «ventaja respiratoria» descrita por Girerd23,24 en las formas asociadas a BMPR2, ausente en la formas idiopáticas y en otras formas hereditarias, lo cual podría reflejar un proceso fisiopatológico distinto en el sistema vascular pulmonar en relación con la vía BMPR2/Smad. No se han observado diferencias significativas en el fenotipo según el tipo de mutación en BMPR2, en contra de lo indicado en otros trabajos8.
Mutaciones en KCNK3Existen hasta la fecha 9 casos de HAP asociada a KCNK3: la serie de Ma et al.13 con 6 pacientes y nuestra serie, con 3 pacientes portadores de 2 variantes patogénicas no descritas previamente. Al tratarse de un gen relacionado hace relativamente poco, es esperable el descubrimiento de nuevas mutaciones en el futuro. Nuestro grupo ha descrito, en un paciente de padres consanguíneos con una forma agresiva de la enfermedad, por primera vez una mutación en homocigosis en KCNK3 (p.Gly106Arg), de la que era portadora la madre afectada. La presencia de ambos alelos mutados en el caso índice podría explicar la agresividad del cuadro, si bien son necesarios futuros estudios para confirmarlo.
Nuestros hallazgos indican, al igual que el trabajo de Ma et al.13, la presencia de un fenotipo más agresivo de las formas asociadas a KCNK3, si bien comparten ciertas características con las formas asociadas a BMPR2, como herencia autosómica dominante, penetrancia incompleta e inicio predominante en la edad adulta. Existen investigaciones dirigidas a restablecer la función de dicho canal25,26, lo cual puede abrir en el futuro una nueva línea de tratamiento en estos pacientes.
Mutaciones en TBX4Además de nuestra serie con 4 pacientes con mutaciones patogénicas en TBX4, solo se ha publicado la serie original de 7 casos de Kerstjens-Frederikse et al.14. Destaca que todos nuestros pacientes con HAP de inicio en la infancia portaban los cambios en el exón 8 y los pacientes con presentación adulta, en el exón 3, fenómeno no observado en la serie de Kerstjens-Frederikse et al.14, en la que los casos de presentación infantil presentaban cambios en ambos exones indistintamente.
Se ha descrito escasamente el fenotipo de este subgrupo de pacientes, aunque se ha señalado que se trata de una forma de presentación predominantemente infantil y de menor agresividad, frecuentemente asociada al síndrome de small patella, si bien ninguno de nuestros pacientes cumple criterios diagnósticos de esta displasia esquelética. Nuestros resultados confirman esos hallazgos, sin ningún evento de muerte o trasplante tras un seguimiento medio de 21,75 años, a pesar de la gravedad hemodinámica basal. La HAP asociada a mutaciones en TBX4 podría representar, por lo tanto, una forma más benigna de la enfermedad, aunque son necesarios más estudios que confirmen esta hipótesis.
Análisis de factores pronósticosEn la población con HAPI, el análisis univariable mostró un impacto pronóstico de los factores clásicos derivados de registros poblacionales como el sexo masculino, la prueba de marcha de 6 minutos y resistencia vascular pulmonar arteriolar, de manera similar a lo publicado previamente27,28, si bien la etiología heredable y la clase funcional de la NYHA basal no presentaron significación, en contra de lo publicado12,27,28;9S,14S del material suplementario (tabla 4). En el análisis multivariable, solo el test de 6 min y el sexo masculino mantuvieron dicho efecto. En el subgrupo de HAPH, el sexo masculino y los parámetros pronósticos clásicos, relacionados con estadios avanzados de la enfermedad (presión de aurícula derecha, resistencia vascular elevada y clase funcional avanzada) mostraron también un impacto negativo en el análisis univariable, aunque solo la clase funcional al diagnóstico mantuvo la significación en el análisis multivariable de este subgrupo. Así, el diagnóstico en clase funcional III o IV de la NYHA en el subgrupo de HAPH se relaciona con un riesgo 3,5 veces superior de entrada en lista de trasplante pulmonar o muerte (tabla 5), hecho previamente no indicado que pone de relevancia la necesidad de un diagnóstico precoz.
Impacto familiarSe ha obtenido un porcentaje significativo de negativas al cribado genético o ecocardiográfico entre los familiares, hecho publicado por otros grupos28. Sin embargo, debido al impacto pronóstico del diagnóstico precoz observado especialmente en las formas hereditarias, parece fundamental lograr un adecuado cribado de familiares y detectar tempranamente a los portadores asintomáticos. De esta manera cobra especial interés el desarrollo de programas de seguimiento de portadores sanos, campo actualmente en intensa investigación28–30.
En cuanto a la penetrancia de las alteraciones descritas, en nuestra población se observa una penetrancia incompleta con gran variabilidad interfamiliar en los 3 genes estudiados, y destacan las marcadas diferencias en función del tipo de mutación en las formas asociadas a BMPR2, si bien la penetrancia general de BMPR2 en nuestra serie es superponible a la publicada previamente6,19. Nuestro grupo es el primero en describir la penetrancia asociada a mutaciones en TBX4, y son necesarios estudios futuros para confirmar nuestros hallazgos.
LimitacionesSi bien el presente estudio ha analizado la mayor muestra española de pacientes con HAP, se debe interpretar con precaución estos hallazgos, fundamentalmente porque se incluyen casos prevalentes e incidentes, por lo que hay una dispersión significativa en cuanto a la fecha del diagnóstico, con el posible sesgo de supervivencia subyacente. Por otro lado, se han estudiado 3 de los genes relacionados con el desarrollo de la enfermedad, incluido BMPR2, el principal gen asociado con la enfermedad, pero se sabe que existen muchos más genes relacionados con la enfermedad que se debería incluir en futuros estudios13,15,16. En cuanto a la categorización de la patogenicidad de las mutaciones encontradas, se trata de una estimación basada en diversos parámetros clínicos y genético-moleculares, y para confirmarla es necesario demostrar cosegregación de la mutación con el fenotipo y/o la realización de estudios funcionales.
CONCLUSIONESEn el presente estudio se ha observado un elemento diferenciador en la población española con HAP con respecto a otras poblaciones descritas, con un porcentaje menor de formas asociadas a BMPR2 y la existencia de un posible y nunca antes descrito punto caliente en el exón 2 de dicho gen. Además, los hallazgos en nuestra población indican que las formas asociadas a TBX4 podrían presentar un fenotipo más benigno, en oposición a las formas asociadas a KCNK3, con un fenotipo más agresivo. No se han observado diferencias en cuanto al pronóstico de las formas idiopáticas frente a las hereditarias, y en nuestra población se mantienen los factores pronósticos de HAP clásicos; destaca el gran impacto pronóstico del diagnóstico precoz en las formas heredables de la enfermedad, con la consiguiente importancia del adecuado cribado, especialmente en este subgrupo de pacientes. Son necesarios nuevos estudios que confirmen estos hallazgos para determinar los matices fenotípicos y pronósticos propios de cada alteración genética en nuestra práctica clínica habitual.
FINANCIACIÓNEste proyecto ha sido parcialmente financiado por la Red de Investigación Cardiovascular del Instituto de Salud Carlos III de Madrid (RD06/0003/0012), así como por becas no condicionadas de la Asociación Española de Hipertensión Pulmonar, Actelion y la Fundación Air Liquide.
CONFLICTO DE INTERESESNinguno.
A la Asociación Española de Hipertensión Pulmonar, Actelion, Foundation Air Liquide, Biobanc del Hospital Clínic de Barcelona y a nuestros pacientes por hacer posible este estudio. Además, los autores manifiestan su agradecimiento a Exome Aggregation Consortium y NHLBI GO Exome Sequencing Project y sus proyectos en marcha —Lung GO Sequencing Project (HL-102923), WHI Sequencing Project (HL-102924), Broad GO Sequencing Project (HL-102925), Seattle GO Sequencing Project (HL-102926) y Heart GO Sequencing Project (HL-103010)— por la valiosa información proporcionada a la comunidad científica.