La caracterización molecular de cardiopatías incluye una entidad congénita no infrecuente, el síndrome de Noonan. Presentamos el estudio de seis genes de la vía RAS-MAPK en pacientes españoles: perfil genotípico, impacto de la cardiopatía y expresividad clínica.
MétodosCompusieron la población en estudio 643 pacientes (y 182 familiares) diagnosticados por dismorfólogos, cardiólogos y endocrinopediatras de 74 hospitales (11 comunidades). Estudio primario de PTPN11 y complementario de SOS1, RAF1, BRAF, KRAS y HRAS, estratificado y orientado por signos clínicos, mediante secuenciación de exones recurrentes (un 80-95% de mutaciones descritas).
ResultadosSe documentó mutación en 230 pacientes (91 mujeres, 139 varones) de 200 familias (31%), 172 PTPN11 +, 14 SOS1 +, 9 RAF1 + y 5 BRAF +, con referencia explícita a la cardiopatía padecida en 156 casos índice; 103 presentaban estenosis de la válvula pulmonar; 12, estenosis de la válvula pulmonar y miocardiopatía hipertrófica; 18, miocardiopatía hipertrófica y 14, otra cardiopatía; en sólo 9 casos se encontraba ausente. En 23/30 familiares positivos no había o no constaba cardiopatía. El rendimiento diagnóstico fue superior (p = 0,016) para las muestras de algunos centros (53%; 14/32), y alcanzó el 64% (9/14; p = 0,019) en profesionales concretos. El rendimiento cayó al 18% en los pacientes sin datos clínicos facilitados. El dato genotípico reorientó el diagnóstico clínico en 26 pacientes.
ConclusionesEl 94% de los pacientes portadores de mutación presentaban cardiopatía, el 79% estenosis de la válvula pulmonar y el 12% miocardiopatía hipertrófica. En el 76% de los familiares positivos con rasgos clínicos compatibles, no se había documentado la cardiopatía. El estudio molecular es una herramienta útil en estos síndromes, aunque debe progresarse en la objetivación del diagnóstico clínico.
Palabras clave
El síndrome de Noonan1 (SN) (OMIM 163950) es un síndrome hereditario considerado relativamente frecuente en la población (1:2.500). Sus signos clínicos son: una dismorfia facial característica, talla baja, cuello ancho y deformidad pectoral, criptorquidia en varones y cardiopatía2, 3, 4 (generalmente estenosis de la válvula pulmonar [EP] y también miocardiopatía hipertrófica [MH]), y se aceptan de manera generalizada los criterios de Van der Burgt5 para su diagnóstico. Es una entidad monogénica dominante que a menudo aparece de novo.
El SN ha pasado de ser interpretado como el «Turner de los varones» a ser reconocido como un síndrome debido a la alteración puntual monoalélica (dominante) de genes autosómicos. Las mutaciones del gen PTPN11 en 12q24.1 que codifica una fosfatasa SHP2 son las primeras alteraciones que se documentaron6 en aproximadamente un 50% de los pacientes. Posteriormente se detectó que algunos pacientes PTPN11 negativos (2-5%) presentaban mutación en KRAS7, 8 (12p12.1), lo que puso en evidencia que las alteraciones de la vía RAS/MAPK podían dar lugar al síndrome. Esta vía reguladora, inicialmente conocida por su implicación en procesos tumorales, controla el equilibrio proliferación/apoptosis y migración celular y, por lo tanto, la morfogénesis. A diferencia de las mutaciones oncogénicas que son de tipo somático y se limitan al clon afectado, la alteración en el síndrome dismórfico ocurre en línea germinal y es compatible con la viabilidad del individuo. Las variantes que causan uno u otro tipo de afección generalmente son distintas8.
Los estudios más recientes han establecido que las alteraciones de otros genes codificantes de las proteínas de la vía RAS/MAPK, las cinasas RAF10, 19 (en 3p25), BRAF11 (en 7q34) y MEK112 (en 15q21) y la proteína intercambiadora de nucleótidos de guanina SOS113, 14 (en 2p22-p21), contribuyen también a la base molecular del SN. El gen SOS1, aunque fue descrito posteriormente, ha pasado a ocupar la segunda posición en cuanto a su contribución al SN, el 20% de los PTPN11 negativos15. Recientemente16 se han documentado también mutaciones en NRAS (1p13.2), aunque con una frecuencia mucho menor (0,4%).
Paralelamente se puso de manifiesto que varios síndromes que mostraban cierto solapamiento fenotípico con el SN, como el LEOPARD17, 18, 19 (OMIM 151100), el cardiofaciocutáneo12, 20 (CFC, OMIM 115150), el de Costello21 (OMIM 218040), la neurofibromatosis tipo I22 (OMIM 162200) y Legius (OMIM 611431), también se deben a alteraciones de esta vía, y se extendía la implicación a HRAS (en 11p15.5), SPRED (en 15q13.2) y MEK2 (en 19p13.3). Todos estos síndromes dismórficos presentaban mutaciones dominantes y recurrentes, y se ha propuesto una denominación común para este grupo de entidades: «síndromes RAS-MAPK», «neurocardiofaciocutáneos» o «rasopatías»23, 24. Existen asociaciones entre algunos signos clínicos y los genes implicados que pueden ayudar a establecer una estratificación del análisis molecular25, 26, 27, 28, 29, 30, 31.
Los objetivos de este trabajo son conocer el perfil genotípico y el rendimiento diagnóstico del análisis molecular en los pacientes españoles con síndrome de Noonan y relacionados y, en quienes han resultado portadores de mutación, valorar el impacto de la cardiopatía y la expresividad clínica.
MétodosPacientesEn este trabajo se incluyen los resultados del análisis molecular realizado entre enero de 2005 y diciembre de 2010 en un total de 643 pacientes (397 varones y 246 mujeres) en quienes se sospechaba SN o síndrome neurocardiofaciocutáneo relacionado (Figura 1). Solicitaron los estudios los especialistas responsables de los pacientes (cardiólogos, endocrinopediatras y dismorfólogos genetistas, y con menor frecuencia neuropediatras y neonatólogos) que realizaron el consejo genético antes y después de la prueba y obtuvieron el consentimiento informado en el hospital de origen. Las muestras procedían de 74 hospitales de 11 comunidades. El formulario preanalítico que se ofreció a todos los centros (material adicional) se basa en el utilizado por Zenker et al27 en el estudio que mostraba la más rigurosa selección fenotípica y la superior caracterización genotípica.

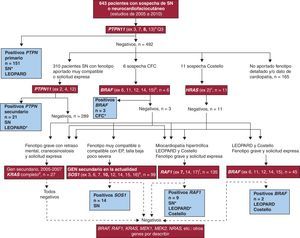
Figura 1. Diagrama de flujo ilustrativo del estudio de genes de la vía RAS-MAPK realizado en los pacientes. Genes estudiados, signos clínicos y/o el síndrome sospechado en los pacientes y número de muestras sometidas a cada estudio en el periodo 2005-2010 recogido en el trabajo. Los genes analizados se enmarcan en recuadro rojo y entre paréntesis se indican los exones recurrentes analizados. En recuadro azul se indica el número de pacientes portadores detectados y los síndromes que presentaban, con asterisco (*) el más frecuente. El estudio primario de PTPN11 se aplicó a todas las muestras; se excluyó de análisis adicionales a los pacientes de los que no se disponía de fenotipo detallado y/o dato de cardiopatía en el periodo cubierto por el estudio, aunque se los contabilizó para el cálculo del rendimiento diagnóstico general del análisis molecular. aEl estudio primario de PTPN11 (4 exones donde se ha documentado el 86% de las mutaciones descritas para este locus6 ) se realizó en todas las muestras, y el secundario (3 exones adicionales, con los que en total se cubre el 99% de las mutaciones descritas en la literatura internacional 25–28 ) se aplicó sistemáticamente a los pacientes con fenotipo disponible y cardiopatía tipo estenosis de la válvula pulmonar o miocardiopatía hipertrófica y en los LEOPARD. Se analizaron otros genes de forma complementaria, como se indica en el diagrama. bBRAF es el gen analizado en los pacientes con síndrome cardiofaciocutáneo y se estudia de forma secundaria en los pacientes con síndrome de Costello, en los LEOPARD negativos para PTPN11 y RAF1.cEl análisis HRAS fue el estudio inicial en los pacientes con síndrome de Costello y se dirigió a las Gly12 y Gly13 que se encuentran mutadas en un 85-90% de estos pacientes 21 . dKRAS fue en una primera etapa el segundo gen a analizar en síndrome de Noon, síndrome de Costello y síndrome cardiofaciocutáneo. Actualmente ha sido desplazado por eSOS1 (síndrome de Noonan) y fRAF1 (pacientes con miocardiopatía hipertrófica, LEOPARD y síndrome de Costello) en nuestros estudios secundarios. CFC: síndrome cardiofaciocutáneo; EP: estenosis de la válvula pulmonar; SN: síndrome de Noonan.
Eran principalmente pacientes pediátricos; el 95% eran menores de 20 años (8,58 ± 17,2 años). Se analizó a 182 familiares en 98 familias (15%). Se encontró mutación en 230 pacientes, 30 de ellos familiares. De 196 pacientes portadores de mutación, se disponía de datos clínicos, con referencia explícita al tipo de cardiopatía padecida en 156 (ausente en 9 de ellos). Con el fin de evitar sesgos en la disponibilidad de datos o hacer más subjetiva la exploración clínica de los pacientes portadores de mutación, se evitó solicitar información una vez obtenido el dato molecular, aunque se nos comunicó que se había modificado el diagnóstico clínico de algunos pacientes.
Para el estudio molecular, se manejaron fundamentalmente dos tipos de muestras, sangre anticoagulada con ácido etilendiaminotetraacético o ADN extraído en el hospital solicitante. En muy pocas ocasiones se analizaron otros tejidos: tejidos renal y hepático de una necropsia de un fallecido a término y cuatro muestras prenatales.
Estudio genético molecularEl estudio molecular se inició en 2005 con el análisis de PTPN11; los genes descritos posteriormente se incorporaron al estudio, como se muestra en la Figura 1, que detalla el proceso de genotipificación. El diagrama no debe interpretarse como un algoritmo definitivo, porque la base molecular de estos síndromes no se conoce totalmente; la serie no es exhaustiva en los genes analizados y sigue abierta a la aportación de datos para la estratificación y caracterización de nuevos casos. En 81 pacientes se analizaron dos genes; en 38, tres; en 15, cuatro; en 6, cinco y en 1, los seis genes. En los 182 familiares, el estudio se dirigió al exón mutado en el paciente índice de la correspondiente familia, para detectar o descartar la alteración. La secuenciación se realizó de forma bidireccional utilizando didesoxinucleótidos fluorescentes y ABI Prism® 3100 (Línea Instrumental de Secuenciación del Hospital General Universitario Gregorio Marañón) analizando los resultados con SeqScape 2.5.
Las alteraciones detectadas no descritas previamente se valoraron in silico mediante: MutPred32 versión 1.2 (Buck Institute, Indiana University; http://mutpred.mutdb.org) y SIFT33 versión 4.0.3 (J. Craig Venter Institute; http://sift.jcvi.org). Estos programas exploran la conservación evolutiva y la funcionalidad de la proteína mutada (deducida a partir de la alteración del plegamiento, la interacción de residuos aminoacídicos neutros o cargados y otras características de los modelos funcionales acordes) y permiten inferir el potencial carácter patogénico de un cambio de secuencia.
Los datos se analizaron mediante el test de comparación de proporciones de Epidat 3.1.
ResultadosEstudios genotípicosEn la Tabla 1 se recogen las alteraciones detectadas en 200 pacientes españoles (126 varones y 74 mujeres) y la cardiopatía que más frecuentemente se encontró asociada. La serie incluye tanto a los pacientes de novo como a los pacientes índice de las formas familiares detectadas. La distribución por sexos de los pacientes portadores de mutación refleja el sesgo hallado en la muestra original. La Figura 1 muestra el diagrama del estudio aplicado y los síndromes asociados a las distintas mutaciones.
Tabla 1. Alteraciones de los genes RAS-MAPK detectadas en 200 pacientes índice con síndrome de Noonan y otros síndromes neurocardiofaciocutáneos. Casos de novo y formas familiares. Genotipo y cardiopatía
| Pacientes índice (n.o de casos) | ||||||||||
| Gen | Dominio proteína | Exón | Genotipo a | Portadores de la alteración | Distribución de frecuencias b | Forma de novo | Forma familiar | Cardiopatía c | Con cardiopatía | Sin cardiopatía |
| PTPN11 | Dominio regulador | 2 | p.Thr42Ala | 4 | 2% | 4 | 0 | EP* | 3 | |
| 3 | p.Thr52Ile | 1 | 1% | 0 | 1 | EP | 1 | |||
| 3 | p.Asn58Asp, p.Asn58His, Asn58Lys | 5 | 3% | 5 | 0 | EP* (3), otra | 4 | |||
| 3 | p.Gly60Ala, p.Gly60Ser | 4 | 2% | 4 | 0 | EP | 2 | 1 | ||
| 3 | p.Asp61Gly, p.Asp61Asn | 8 | 5% | 8 | 0 | EP* (5), otra | 7 | 1 | ||
| 3 | p.Tyr62Asp | 2 | 1% | 2 | 0 | EP* (1), otra | 2 | |||
| 3 | p.Tyr63Cys d | 10 | 6% | 7 | 3 | EP* (5), otra | 6 | |||
| 3 | p.Ala72Gly, p.Ala72Ser | 9 | 5% | 9 | 0 | EP* (6), MH | 8 | |||
| 3 | p.Thr73Ile, p.Thr73Leu | 3 | 2% | 3 | 0 | EP* | 1 | 1 | ||
| 3 | p.Glu76Asp | 1 | 1% | 1 | 0 | |||||
| 3 | p.Gln79Arg | 9 | 5% | 7 | 2 | EP (5) | 5 | |||
| 3 | p.Asp106Ala | 3 | 2% | 3 | 0 | EP* | 3 | |||
| Región puente | 4 | p.Glu110Lys | 1 | 1% | 1 | 0 | ||||
| 4 | p.Glu139Asp | 6 | 3% | 6 | 0 | EP* | 5 | 1 | ||
| Dominio centro activo | 7 | insCAA(Gln256) e | 1 | 1% | 0 | 1 | ||||
| 7 | p.Gln256Arg | 1 | 1% | 1 | 0 | EP | 1 | |||
| 7 | p.Leu261Phe | 1 | 1% | 1 | 0 | |||||
| 7 | p.Gly268Cys | 1 | 1% | 1 | 0 | 1 | ||||
| 7 | p.Tyr279Cys | 5 | 3% | 4 | 1 | EP (1), MH | 2 | |||
| 7 | p.Ile282Val | 3 | 2% | 3 | 0 | EP* (1), otra | 2 | |||
| 7 | p.Phe285Leu, | 5 | 3% | 4 | 1 | EP* (2), otra | 3 | |||
| 8 | p.Phe285Cys, p.Phe285Ser | 3 | 2% | 3 | 0 | EP* | 2 | |||
| 8 | p.Asn308Asp, p.Asn308Ser | 56 | 33% | 53 | 3 | EP* (33), MH, otra | 37 | 3 | ||
| 12 | p.Ala461Ser, p.Ala461Thr | 2 | 1% | 2 | 0 | MH* | 2 | |||
| 12 | p.Thr468Met | 9 | 5% | 8 | 1 | EP* (5), MH* | 9 | |||
| 13 | p.Pro491Ser, p.Pro491Thr | 4 | 2% | 3 | 1 | EP* | 3 | |||
| 13 | p.Ser502Ala, p.Ser502Leu | 2 | 1% | 2 | 0 | EP* | 2 | |||
| 13 | p.Gly503Arg | 1 | 1% | 1 | 0 | EP* | 1 | |||
| 13 | p.Met504Val | 10 | 6% | 8 | 2 | EP* (5), MH | 7 | |||
| 13 | p.Gln510Glu, p.Gln510Pro, p.Gln510Arg | 3 | 2% | 2 | 1 | EP (1), MH | 2 | 1 | ||
| SOS1 | Dominio DH | 6 | p.Thr266Lys | 2 | 11 | 3 | EP* | 2 | ||
| 6 | p.Met269Thr | 2 | EP* | 2 | ||||||
| 7 | p.Asp309Tyr | 1 | otra | 1 | ||||||
| Dominio PH | 10 | p.Trp432Arg | 2 | EP* | 2 | |||||
| Dominio unión PH-REM | 10 | p.Ser548Arg | 1 | EP | 1 | |||||
| 10 | p.Arg552Ser, p.Arg552Gly, p.Arg552Trp | 5 | EP* (4) | 5 | ||||||
| 10 | c.1330_1332del f | 1 | EP | 1 | ||||||
| RAF1 | Dominio CR2 | 7 | p.Ser257Leu | 5 | 9 | 0 | MH* | 5 | ||
| 7 | p.Ser259Phe | 1 | MH | 1 | ||||||
| 7 | p.Pro261His, p.Pro261Thr | 2 | MH* | 2 | ||||||
| Dominio activación (CR3) | 14 | p.Glu478Lys | 1 | MH | 1 | |||||
| BRAF | CRD (CR1) | 6 | p.Gln257Arg | 2 | 5 | 0 | MH, otra | 2 | ||
| Dominio activador (CR3) | 12 | p.Glu501Lys | 2 | EP* | 2 | |||||
| 14 | p.Asn581Asp | 1 | ||||||||
| 180 | 20 | 147 | 9 | |||||||
| Totales | 200 | 200 | 156 | |||||||
EP: estenosis de la válvula pulmonar; MH: miocardiopatía hipertrófica.
a Las alteraciones detectadas en los pacientes españoles eran, con excepcionese,f, mutaciones ya documentadas en pacientes con síndrome de Noonan u otros neurofaciocutáneos en otras poblaciones. No se incluyen las variantes intrónicas ni los polimorfismos descritos en zonas codificantes. En la figura se indican los tipos de síndromes que presentaron los pacientes portadores de las mutaciones de los distintos genes.
b Frecuencia de las alteraciones en los distintos dominios de la proteína codificada por el gen PTPN11. No se incluyen los porcentajes para los demás genes, dado el número reducido de alelos portadores.
c Se señala con * el tipo de cardiopatía que se encontraba presente o resultó predominante en los pacientes portadores de la alteración indicada en la columna de genotipos. Cuando se encontró asociado más de un tipo de cardiopatía, se indica entre paréntesis el número de pacientes con estenosis de la válvula pulmonar (EP).
d Uno de los pacientes que presentó la mutación p.Tyr63Cys mostró una alteración adicional del exón 8, p.Met311Val en cis con la mutación, que cosegregaba en los familiares que presentaban la enfermedad (3 casos). La variante p.Met311Val no ha sido descrita y, aunque no se ha detectado en 700 cromosomas normales (analizados mediante secuenciación parcial de PTPN11), los estudios in silico32,33 (véase «Métodos») indican que se trata únicamente de un polimorfismo.
e En una forma familiar se caracterizó una alteración nueva en PTPN11 que cosegregaba con el fenotipo e implicaba la inserción en fase del triplete CAA en el exón 7. Este triplete codificante del aminoácido glutamina (Gln) se localiza adyacente al residuo Gln256 (ins CAA, Gln256) y no se ha detectado en los 700 cromosomas normales analizados.
f Un paciente de novo presentó la alteración de SOS1 c.1330_1332del. Esta alteración no descrita se ha descartado en 1.000 cromosomas normales mediante cribado HRM (high resolution melting), y se encuentra en preparación el manuscrito que la describe.
La tasa de caracterización obtenida para el estudio de PTPN11 fue del 27% (172/643 pacientes). El rendimiento diagnóstico de PTPN11 se incrementa al 43% (128/292) si consideramos únicamente el grupo de pacientes de los que constaba la existencia de cardiopatía. Sólo 9/129 (7%) pacientes PTPN11 positivos no presentaban cardiopatía.
La distribución de la frecuencia de mutaciones fue del 34% en zona reguladora, el 4% en zona puente y el 62% en zona dominio fosfatasa. Se detectaron formas de novo y familiares en ambas zonas, reguladora y centro activo (Tabla 1). La alteración más recurrente, p.Asn308Asp (n = 47), se localiza en el dominio fosfatasa, aunque también se detectó el cambio p.Asn308Ser (n = 9). En total, 107 alelos mutados se localizaron en este dominio. Las mutaciones de la región reguladora del centro activo constituyen el segundo bloque mayoritario (59 pacientes). El perfil fue más heterogéneo con 12 residuos implicados, aunque afectan primordialmente al exón 3.
KRASEl estudio del gen KRAS se planteó inicialmente como primera alternativa en los casos PTPN11 negativos con sospecha clínica firme, aunque no se caracterizaron alteraciones en ningún caso. Actualmente se aplica como estudio de tercer o cuarto nivel para fenotipos severos que asocian retraso mental.
SOS1En el periodo recogido en este estudio se caracterizó a 14 pacientes, todos con diagnóstico de SN que asociaba cardiopatía, primordialmente EP pero también ocasionalmente MH. El exón 10 mostró una alta recurrencia, concretamente la alteración del residuo arginina 552 se detectó en 5 pacientes, con tres tipos de cambios de aminoácido p.Arg552Gly, p.Arg552Ser y p.Arg552Trp. En esta región hemos tenido ocasión de detectar una alteración de novo no descrita previamente: p.Asp430del (c.1330_1332del, anteriormente c.1330_1332delATG). Se trata de una deleción «en fase» que implica a un aminoácido fuertemente conservado: aspartato 430 (manuscrito en preparación). En tres de las 14 familias con mutación en SOS1 se detectó a familiares afectados.
RAF1En esta serie 9 pacientes presentaron mutación en RAF1; la alteración más recurrente (5 casos) fue la p.Ser257Leu. Cabe reseñar que la mutación detectada fuera de este exón, la p.Asp478Lys del exón 14, es la única en esta serie que había sido descrita en tumores esporádicos como mutación somática de novo9. Se confirmó que los padres asintomáticos no presentaban la alteración, por lo que la mutación se había producido de novo en línea germinal. En 2 pacientes no relacionados y procedentes de distintos hospitales con sospecha de Costello, se detectó la mutación p.Ser257Leu del gen RAF1 (casos 6 y 9; Tabla 2).
Tabla 2. Pacientes y familiares de síndromes RAS-MAPK en los que el diagnóstico molecular reorientó el diagnóstico clínico
| Caso a | Familia | PacientesCasos índice | Diagnóstico inicial | Genotipo | Diagnóstico final |
| 1 | 1 | Índice | Noonan | PTPN11-p.Thr468Met | LEOPARD |
| 2 | 2 | Índice | Neurofibromatosis tipo I con MH | PTPN11-p.Thr468Met | LEOPARD 16 |
| 3 | 3 | Índice | Neurofibromatosis tipo I con MH | PTPN11-p.Thr468Met | LEOPARD 16 |
| 4 | 4 | Índice | Neurofibromatosis tipo I con MH | PTPN11-p.Thr468Met | LEOPARD 16 |
| 5 | 5 | Índice | Neurofibromatosis tipo I con MH | PTPN11-p.Thr468Met | LEOPARD |
| 6 | 6 | Índice | Costello c | RAF1-p.Ser257Leu | Noonan con MH |
| 7 | 7 | Índice | Noonan | BRAF-p.GlnQ257Arg | CFC |
| 8 | 8 | Índice | Cardiopatía, sin diagnóstico sindrómico | BRAF-p.GlnQ257Arg | LEOPARD |
| 9 | 9 | Índice | Costello c | RAF1-p.Ser257Leu | Noonan con MH |
| Familiares | |||||
| 10 | 10 | Madre | LEOPARD familiar | PTPN11-normal | No afectada, efélides muy abundantes |
| 11 | 10 | Afectado (índice) | LEOPARD familiar | PTPN11-p.Asn308Asp | Noonan de novo |
| 12 | 11 | Madre (índice) | Talla baja y facies sugestiva no diagnosticada | PTPN11-p.Tyr63Cys | Noonan |
| 13 | 11 | Hijo b | Hidrops faetalis (muerte neonatal) | PTPN-p.Tyr63Cys | Noonan |
| 14 | 12 | Madre | Talla baja y pterigium coli (sospecha de Turner) | PTPN-p.Asn308Asp | Noonan |
| 15 | 13 | Padre | Critorquidia | PTPN-p.Pro491Thr | Noonan |
| 16 | 14 | Padre | Criptorquidia bilateral | SOS1-p.Arg552Trp | Noonan |
| 17 | 15 | Madre | Talla baja y fenotipo levemente sugestivo | PTPN11-normal | No afectada |
| 18 a 26 | Otros familiares d | No consulta, clínica actual en evaluación | 7 PTPN+ y 2 SOS1+ | Noonan |
CFC: síndrome cardiofaciocutáneo; MH: miocardiopatía hipertrófica.
a Los casos recogidos en esta tabla son exclusivamente aquellos cuyo cambio de diagnóstico nos consta, por lo que la selección puede no ser exhaustiva.
b Estudio de necropsia, tejidos renal y hepático.
c Dos pacientes no relacionados procedentes de distintos hospitales con sospecha de síndrome de Costello presentaron la mutación p.Ser257Leu del gen RAF1.
d Nueve familiares con genotipo positivo de los que no se consignó que tuvieran signos clínicos.
El estudio de HRAS aplicado a los pacientes con sospecha de síndrome de Costello descartó las alteraciones recurrentes en todos los analizados en el periodo cubierto por este estudio. En 3 pacientes con esta sospecha diagnóstica se caracterizaron mutaciones en alguno de los genes analizados (Figura 1).
BRAFSe detectó a 5 pacientes BRAF positivos. En 3 de ellos, se confirmó el diagnóstico clínico original de CFC; en los otros 2, se reorientó por el hallazgo molecular (Tabla 2; casos 7 y 8).
La suma de las tasas de caracterización obtenidas para cada uno de los exones analizados alcanza un 63% en los estudios estratificados por fenotipo, acorde con la franja descrita en la literatura. El incremento del número de genes analizados mejora la caracterización, aunque de forma cada vez menos eficiente: el 20% (16/81), el 13% (5/38) y el 6% (1/15) con 2, 3 y 4 genes. En el grupo analizado para 5 genes (6 casos), sin embargo, se caracterizó a 2 pacientes, aunque uno resultó positivo para SOS1, que actualmente ha desplazado a KRAS y se analiza como segunda opción del análisis molecular.
Relaciones genotipo-fenotipoTodos los genes y todas las alteraciones moleculares asociaron tanto EP como MH en unos u otros pacientes (Tabla 1), aunque la EP aparecía en todas ellas y la MH se encontró más limitada (aunque no exclusivamente) a RAF1 y a la mutación concreta de PTPN11 p.Thr468Met. La cardiopatía más relacionada con la recurrente alteración de PTPN11 p.Asn308Asp fue la EP (33 de los 40 pacientes con signos informados), sólo 1 paciente presentaba MH y en 3 no había cardiopatía. Las mutaciones en RAF1 se observaron más frecuentemente en los pacientes con MH (9/40 con MH frente a 0/28 con EP) y en el LEOPARD (2/12). La mutación p.Thr468Met asoció muy fuertemente el fenotipo LEOPARD y, de hecho, en algunos de los pacientes el dato molecular anticipó el diagnóstico clínico del LEOPARD, ya que las manchas café con leche y lentigos típicos aparecieron posteriormente (Tabla 2; caso 1). Curiosamente aunque el acrónimo dirige la atención a la EP, en nuestra serie cerca de la mitad de los pacientes con esta sospecha diagnóstica han mostrado MH. En 3 pacientes referidos por Carcavilla et al19, el diagnóstico inicial había sido de neurofibromatosis tipo I.
Las alteraciones hematológicas se encontraron primordialmente asociadas al exón 3 de PTPN11; algunos eran diagnósticos neonatales30. Sin embargo, en ninguno de nuestros pacientes con mutación del aminoácido típicamente asociado25 Thr73 se habían manifestado estos procesos; aunque todos eran pacientes pediátricos.
El solapamiento genotípico y fenotípico de los síndromes neurocardiofaciocutáneos puede observarse en la Figura 1, donde se indica para cada gen el número de pacientes que padecían los distintos síndromes. En al menos 9 pacientes índice, la mutación identificada reorientó el diagnóstico de sospecha por el que los habían remitido (Tabla 2).
Estudios familiaresLas formas familiares se detectaron en pacientes portadores de mutaciones en PTPN11 (n = 17; 11 en la región codificante del dominio centro activo y 6 en la región reguladora) y en SOS1 (n = 3) (Tabla 1). Se detectaron en total 30 casos de familiares portadores de mutación en estas 20 familias. La herencia fue más frecuentemente materna: 14 casos índice pediátricos cuya madre resultó portadora y 1 paciente adulta cuya hija resultó afectada. En un paciente índice la sospecha diagnóstica en la madre fue consecuencia de una muerte perinatal. La confirmación molecular en la madre precedió al estudio post mortem que confirmó el diagnóstico (Tabla 2; caso 13). Los demás casos familiares detectados correspondieron a 7 hermanos en 6 familias, un tío y un abuelo, ambos maternos. En una forma familiar (madre e hijo portadores), el estudio de los abuelos pudo documentar que la mutación se había producido de novo en esa generación previa.
La cardiopatía, que como hemos visto es un hallazgo casi constante en los casos índice portadores de mutación, se encontraba muy infrecuentemente diagnosticada y/o valorada en los progenitores que presentaron la alteración. En 4 casos había signos clínicos compatibles que no habían llevado al diagnóstico (talla baja, criptorquidia) y en 9 casos no había habido consulta, diagnóstico o evaluación previa al hallazgo molecular (Tabla 2). Sólo en 3 de los 19 adultos positivos detectados en el estudio familiar, la cardiopatía se había diagnosticado previamente.
Rendimiento diagnóstico por centros y facultativosEl rendimiento diagnóstico obtenido no fue homogéneo y se observaron diferencias entre los distintos hospitales solicitantes. En la Tabla 3 se recogen los datos relativos a los centros con un número de estudios solicitados no inferior a 9 pacientes (tasa máxima del 54%). Aunque no se muestra la caracterización desglosada por profesionales, sí debemos reseñar que para algún facultativo superó este porcentaje (9/14, 64,3%; p = 0,01).
Tabla 3. Rendimiento diagnóstico del estudio molecular obtenido para las muestras de pacientes procedentes de una selección de hospitales del ámbito nacional con un número de solicitudes no inferior a 9 en el periodo analizado 2005-2010
| Hospital a | Estudios solicitados b | Sólo pacientes índice | Casos positivos c | Pacientes índice positivos | Rendimiento d |
| 1 | 137 | 105 | 34 | 30 | 28,6% |
| 2 | 100 | 70 | 19 | 15 | 21,4% |
| 3 | 51 | 49 | 17 | 17 | 34,7% |
| 4 | 53 | 44 | 13 | 13 | 29,5% |
| 5 | 46 | 44 | 12 | 11 | 25% |
| 6 | 55 | 32 | 20 | 17 | 53,1% |
| 7 | 33 | 30 | 8 | 6 | 20% |
| 8 | 29 | 26 | 11 | 11 | 42,3% |
| 9 | 28 | 25 | 2 | 2 | 8% |
| 10 | 31 | 24 | 4 | 4 | 16,7% |
| 11 | 26 | 22 | 9 | 9 | 40,9% |
| 12 | 35 | 19 | 11 | 7 | 36,8% |
| 13 | 30 | 19 | 9 | 9 | 47,4% |
| 14 | 20 | 17 | 3 | 2 | 11,8% |
| 15 | 18 | 15 | 6 | 5 | 33,3% |
| 16 | 16 | 13 | 4 | 5 | 38,5% |
| 17 | 12 | 11 | 5 | 5 | 45,5% |
| 18 | 9 | 6 | 4 | 3 | 50% |
| Total | 571 | 171 | 34,0% |
a Ordenados por el número de estudios solicitados en el periodo 2005-2010 desde el centro en cuestión.
b Incluye todas las muestras analizadas, pacientes índice y familiares afectados y no afectados.
c Se refiere al total de pacientes y casos familiares portadores de mutación.
d El rendimiento se calcula a partir del número de pacientes índice analizados y caracterizados (positivos) debido a que los estudios de los ADN parentales, solicitados por consejo genético en ausencia de un fenotipo sugestivo, frecuentemente son negativos en ambos progenitores. El rendimiento resulta discretamente superior al general, lo que quizá refleje la mayor experiencia derivada de la casuística más amplia de estos centros.
El rendimiento general del estudio molecular (200/643; 31%) se incrementa al 50% (147/292) si nos ceñimos a los pacientes con cardiopatía, y al 62% (134/218) si esta era del tipo EP y/o MH. Cabe destacar que el tipo de cardiopatía en los pacientes portadores de mutación fue mayoritariamente EP y/o MH (139/156; 89%), mientras que el espectro de la cardiopatía se diversificaba y este porcentaje resultaba menor en los pacientes que no presentaban mutación (84/147 [54%] con EP y/o MH; p < 0,0001). La ausencia de cardiopatía, una rareza en el grupo de pacientes portadores de mutación (9/156), resultó menos infrecuente en el grupo de pacientes negativos (101/343; p < 0,0001).
Se observó un menor rendimiento diagnóstico en el grupo de pacientes de cuyos datos fenotípicos no se disponía (el 18 frente al 31%; p = 0,0002). La exclusión de pacientes sin información relativa a la cardiopatía incrementaba el rendimiento diagnóstico (156/400 [39%]; p = 0,01).
DiscusiónLa caracterización molecular de 230 pacientes españoles con SN u otros síndromes RAS-MAPK ha permitido la confirmación del diagnóstico clínico y ha facilitado un diagnóstico diferencial más preciso. Esta clasificación más efectiva apoyada en la evaluación genómica puede facilitar unos tratamientos mejor orientados, como señalaba Rodríguez-Marín34.
Las alteraciones detectadas en nuestra población han sido en su gran mayoría mutaciones ya descritas, lo que facilitó la elaboración de los informes de diagnóstico. La distribución de las mutaciones fue similar a lo publicado sobre otras poblaciones25, 26, 27, 28. Las alteraciones en algunos aminoácidos, PTPN11-Asn308, SOS1-Arg552 y RAF1-Ser257, fueron recurrentes.
El perfil genotípico observado no difiere del descrito en otras poblaciones: PTPN11 es el gen mayoritario y SOS1 ocupa la segunda posición. La asociación de MH hace primordial el estudio de RAF1 en los pacientes PTPN11 negativos, y cuando hay rasgos de CFC, el análisis primario de BRAF. Sin embargo, no debe descartarse el análisis complementario de estos genes en pacientes con sospecha de otros síndromes RAS-MAPK; de hecho, en 2 casos no relacionados con sospecha de Costello, generalmente asociado a HRAS21, se detectó una misma mutación de RAF1. El diagnóstico diferencial se vio facilitado muy especialmente en el periodo neonatal30 y en el LEOPARD19. El estudio estratificado planteado es acorde con las recomendaciones actuales de Romano et al29, que proponen un abordaje estratificado basado en el fenotipo frente a genotipificación mediante arrays.
El diagnóstico molecular ha permitido descartar el síndrome en algunos familiares que compartían signos pero no padecían la enfermedad y pudo confirmar post mortem una forma familiar. No menos importante es que se detectó a familiares afectados con cardiopatía no reconocidos previamente. Cabe destacar que en dos progenitores portadores de mutación, el único signo recogido en las historias era la criptorquidia. Como se ha demostrado recientemente35, la supervivencia de los pacientes con SN y MH se encuentra significativamente reducida frente a los pacientes con MH no sindrómica, y en ocasiones la expresividad facial no facilita la detección de afectados19, lo que hace del análisis molecular una herramienta diagnóstica imprescindible. Recientemente Sánchez et al3 han señalado la asociación del SN y la MH.
En los pacientes con cardiopatía tipo EP o MH, el rendimiento diagnóstico se ha visto muy favorecido, el 62 frente al 31% general. La tasa de caracterización ha sido extremadamente variable para los distintos centros, aun excluyendo aquellos con un número de estudios reducido. El rendimiento diagnóstico general, que era similar al descrito en series carentes de selección rigurosa26, alcanzó el 64% en profesionales concretos, lo que indica que el abordaje molecular planteado es adecuado y que se debe mejorar la forma de establecer la sospecha diagnóstica en nuestro medio.
Los factores que han favorecido la mayor tasa de caracterización genotípica son, aparte de la mera aportación de datos clínicos, la evaluación del paciente por dismorfólogos expertos y la existencia de cardiopatía. Los criterios clínicos de Van der Burgt5, ampliamente aceptados y utilizados, otorgan un gran protagonismo a la facies característica como clave diagnóstica. Nuestros datos en pacientes con genotipo positivo dan relevancia a la existencia y el tipo de cardiopatía que, quizá por ser un criterio más objetivo y uniforme, en nuestro medio es un dato que ha orientado con más especificidad la sospecha diagnóstica.
La cardiopatía es a veces el motivo de primera consulta en los pacientes con SN, y es primordial que los cardiólogos puedan sospecharla y estén sensibilizados ante un síndrome hereditario que frecuentemente aparece de novo, por lo que no se dispondrá del fenotipo familiar para apoyar la sospecha. Resulta esencial disponer de pruebas complementarias adecuadas y los estudios moleculares han demostrado que pueden facilitar la confirmación diagnóstica y la ayuda a los familiares. Estamos trabajando en la optimización de otros marcadores que puedan contribuir a objetivar la sospecha, como la morfometría en fotos faciales36 o la detección de la sobrexpresión de mensajeros37 de la vía RAS-MAPK.
Las limitaciones del estudio se derivan principalmente de la procedencia diversa de los pacientes y de la escasez de datos clínicos considerados. Su principal fortaleza reside en el número de pacientes estudiados. La obtención exhaustiva de datos clínicos que permitiría hacer un estudio de correlación genotipo/fenotipo está fuera de las pretensiones de este estudio. Por otra parte, la selección de pacientes con datos detallados podría haber sesgado los resultados obtenidos, por lo que entendemos que nuestro enfoque permite obtener una imagen más real de la situación en nuestro medio.
ConclusionesHay cierta heterogeneidad en la forma en que se plantea la sospecha diagnóstica de los síndromes neurocardiofaciocutáneos en nuestro medio y el dato molecular ha sido clave para diagnosticar mejor y detectar los casos familiares. El alto grado de asociación de la cardiopatía documentado, unido a la constatación de casos en los que no se había realizado el estudio cardiológico, nos alerta sobre la importancia de que se realicen en los pacientes y familiares los estudios y las exploraciones oportunas.
FinanciaciónFinanciado por el Fondo de Investigaciones Sanitarias (PI 06/1179).
Conflicto de interesesNinguno.
Agradecimientos
Los autores agradecen la colaboración de los pacientes y sus familiares, así como a los distintos servicios y unidades que nos han confiado sus muestras. En especial queremos mencionar a los doctores: Zapico (Hospital de Alicante), Lapunzina (Hospital La Paz), Lopez-Siguero (Hospital Carlos Haya), Del Campo (Hospital Vall d’Hebron), Tamarit y Pozo (Hospital Niño Jesús), Rodrigo-Palacios (Hospital General Yagüe), Galán (Hospital Perpetuo Socorro), Guitart (Hospital Parc Taulí), Martínez (Hospital Reina Sofía), García-Cuartero y Carrasco (Hospital Severo Ochoa), Luzuriaga (Hospital Universitario Marqués de Valdecilla), Lautre (Hospital Clínico San Carlos), Herrera (Complejo Hospitalario de Jaén) y Albiñana (Hospital de Almansa), cuyos casos han sido, por su número o sus particularidades, especialmente relevantes para el interés de este manuscrito. Begoña Ezquieta es investigadora adscrita de la U753 de CIBERER (Centro de Investigación Biomédica en Red de Enfermedades Raras).
Appendix A. Material adicionalSe puede consultar material adicional a este artículo en su versión electrónica disponible en doi:10.1016/j.recesp.2011.12.016.
Anexo A. Material suplementarioRecibido 11 Julio 2011
Aceptado 14 Diciembre 2011
Autor para correspondencia: Laboratorio de Diagnóstico Molecular, Hospital Infantil, Hospital General Universitario Gregorio Marañón, Dr. Esquerdo 46, 28007 Madrid, España. bezquieta.hgugm@salud.madrid.org