La amiloidosis cardiaca es una enfermedad de difícil diagnóstico y tratamiento. Entre los subtipos de amiloidosis cardiacas figuran las formas hereditarias, de las que la causada por mutaciones en el gen de la transtiretina es la más frecuente.
La correcta identificación de los pacientes cuya amiloidosis se debe a un defecto genético tiene gran importancia, ya que modifica la actitud diagnóstica y terapéutica que adoptar con el enfermo y sus familiares.
En este trabajo describimos nuestra experiencia en la evaluación de dos familias con amiloidosis cardiaca hereditaria por transtiretina. Discutimos diversos aspectos relacionados con el abordaje diagnóstico de los pacientes, así como la actitud diagnóstica y terapéutica que adoptar con los familiares.
Palabras clave
La amiloidosis cardiaca es una enfermedad causada por el depósito de un material proteináceo insoluble, la sustancia amiloide1, 2. Esta sustancia anómala puede tener diversos orígenes y composición, que dan lugar a los diferentes subtipos de amiloidosis, cada uno con diferente pronóstico y tratamiento específico1, 2.
Entre los subtipos de amiloidosis se encuentran las amiloidosis hereditarias, en las que el depósito se produce por una alteración genética. De las distintas amiloidosis hereditarias, sólo afectan al corazón la apolipoproteína A-I (AL) y la transtiretina (TTR), y esta es mucho más frecuente1.
La TTR es una proteína de síntesis hepática en cuyo gen se han descrito más de 100 mutaciones. Las diferentes mutaciones pueden dar lugar a diferentes fenotipos, y se han descrito formas neuropáticas, cardiacas, renales y oculares1, 2.
Dada la naturaleza hereditaria de muchas cardiopatías y la posibilidad actual de realizar tests genéticos, el cardiólogo debe incorporar en su práctica la sospecha de estas etiologías. El manejo apropiado de los resultados genéticos tendrá trascendencia en cuanto al consejo genético y la toma de decisiones en el diagnóstico, el seguimiento y el tratamiento del paciente y sus familiares.
MétodosDescribimos nuestra experiencia en el diagnóstico y la evaluación de dos familias con amiloidosis cardiaca hereditaria por TTR.
ResultadosFamilia 1Sujeto índiceVarón de 52 años con antecedentes de hipertensión arterial, hipercolesterolemia y síndrome del túnel carpiano (STC) bilateral intervenido3. A los 48 años requirió un marcapasos por fibrilación auricular y respuesta ventricular lenta. Dos años más tarde, por un cuadro de insuficiencia cardiaca, se realizó un ecocardiograma, que objetivó una miocardiopatía restrictiva compatible con enfermedad de depósito. La biopsia endomiocárdica mostró depósitos de material hialino positivo para rojo Congo.
Pese a la realización de electroforesis e inmunofijación en sangre y orina y biopsia de médula ósea, que fueron normales, la amiloidosis se consideró de tipo AL, y se decidió tratamiento médico expectante.
Durante los siguientes 2 años el paciente sufrió deterioro progresivo de su capacidad funcional, con ingresos frecuentes por insuficiencia cardiaca y disminución de la fracción de eyección, por lo que fue remitido para valoración de trasplante cardiaco (TxC).
La clínica, poco típica de amiloidosis AL por su lenta evolución, la ausencia de pico monoclonal en sangre y orina y una biopsia de médula ósea normal hicieron sospechar un diagnóstico alternativo. La gammagrafía con 99mTc-3,3-difosfo-1,2-propanodicarboxílico (99mTc-DPD) mostró captación en el miocardio, y la inmunohistoquímica sobre muestra cardiaca mostró depósitos de TTR (Figura 1). El estudio genético identificó una mutación heterocigota Glu89Lys en el gen TTR, ya descrita previamente4. Un estudio electroneurográfico (ENG) evidenció una polineuropatía sensitivomotora de grado leve-moderado.

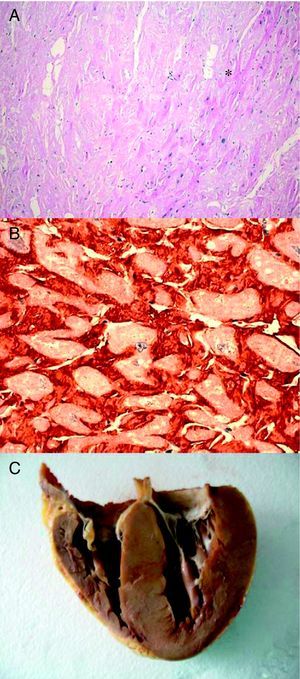
Figura 1. Biopsia endomiocárdica de un paciente con amiloidosis cardiaca hereditaria por transtiretina. A: hematoxilina-eosina. El amiloide (*) aparece como material amorfo entre los miocitos. B: inmunohistoquímica para detección de transtiretina perimiocítica. C: corazón del paciente índice 2. Cortesía de la Dra. Clara Salas Antón.
Ante la mala evolución clínica y considerando que no había afección significativa de otro sistema, se propuso TxC, que se realizó sin complicaciones. Seis meses después fue incluido en lista de trasplante hepático (TxH), donde permaneció 18 meses, tiempo durante el cual no se detectó amiloide en las biopsias endomiocárdicas y no se produjo progresión significativa de la neuropatía. Cuatro años después del TxC y 2 años tras el TxH, el paciente lleva una vida normal.
Evaluación familiarNo existían antecedentes familiares de cardiopatía o muerte súbita. Su madre falleció a los 51 años por tuberculosis y su padre seguía vivo con 72 años sin afecciones significativas.
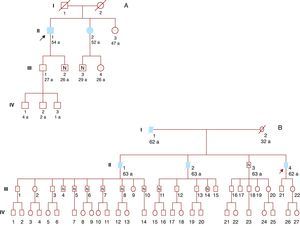
Se ofreció realizar estudio genético a los familiares mayores de 16 años que lo desearan. Se estudió a 6 familiares y encontramos que 4 eran portadores del mismo defecto genético (Figura 2A).

Figura 2. A: familia 1. B: familia 2. Símbolos rellenos: afectados. Símbolos blancos: sin cardiopatía. Símbolos con punto: portadores sin cardiopatía. Símbolos con N: no portadores. Símbolos con línea diagonal: fallecidos. Flecha: paciente índice.
A todos los portadores se les recomendó seguimiento clínico anual y realización periódica de estudios de imagen cardiaca y ENG.
En nuestro centro sólo se sigue al sujeto III:1 que, tras casi 5 años, no presenta signos clínicos de neuropatía/cardiopatía. En su última evaluación, no presentaba depósitos de amiloide en la resonancia magnética (RM) cardiaca ni afección neuropática en el ENG.
Al paciente se le ha dado el pertinente consejo genético. Sus 3 hijos, por ser menores de edad, no se han sometido a estudio genético.
De los demás familiares portadores de la mutación, tenemos constancia que una de las hermanas del sujeto índice, con antecedentes de STC, ha sufrido insuficiencia cardiaca a los 52 años. Pese a nuestra recomendación, ha declinado ser valorada en algún centro con posibilidad de realizar TxC y TxH.
Familia 2Sujeto índiceVarón de 62 años con antecedentes de hipertensión arterial, diabetes mellitus y STC bilateral. Comenzó con síntomas de insuficiencia cardiaca que se interpretaron como secundarios a cardiopatía hipertensiva, hasta que un ecocardiograma evidenció signos de miocardiopatía restrictiva de aspecto infiltrativo. Se realizó biopsia rectal, que mostró depósitos rojo Congo que, mediante inmunohistoquímica, se identificaron como TTR.
Un ENG evidenció polineuropatía sensitivomotora de grado moderado. Tras deterioro de la clase funcional, se remitió para evaluación.
El estudio pronóstico de la cardiopatía mostró parámetros desfavorables y el cateterismo derecho objetivó elevación severa de las presiones pulmonares y gasto cardiaco disminuido. En la RM se observó captación panmural de gadolinio en el miocardio. Una gammagrafía 99mTc-DPD mostró captación cardiaca.
El estudio genético evidenció una mutación heterocigota ya descrita, consistente en la deleción del codón V122 de TTR.
Ante la presencia de resistencias pulmonares elevadas que contraindicaban el TxC, se decidió realizar tratamiento con sildenafilo y bosentán simultáneamente. Tras 3 meses de tratamiento, que fue bien tolerado, se realizó nuevo cateterismo derecho, que mostró reducción de las resistencias pulmonares. Ante la clase funcional avanzada y la necesidad de ingresos frecuentes, se decidió plantear TxC seguido de TxH en un segundo tiempo.
El TxC cursó con insuficiencia ventricular derecha precoz que requirió de implantación de asistencia ventricular derecha. Tras mejoría progresiva y retirada de la asistencia ventricular (28 días tras el TxC), el paciente sufrió múltiples complicaciones infecciosas derivadas de la prolongada estancia en la unidad de cuidados intensivos y falleció a los 50 días del TxC.
Evaluación familiarEl padre del sujeto índice había fallecido a los 62 años por insuficiencia cardiaca. Dos de sus hermanos habían fallecido a los 63 años tras sufrir insuficiencia cardiaca progresiva (uno, por muerte súbita). En el sujeto II:2 se llegó a establecer el diagnóstico de amiloidosis, aunque el subtipo no fue tipificado.
Diecisiete familiares se sometieron a estudio genético y 9 presentaron la mutación (Figura 2B). El único hermano vivo del paciente índice no era portador, por lo que no fue necesario estudiar a sus descendientes. Varios de los descendientes de sus hermanos fallecidos presentaban la mutación, por lo que estos eran portadores obligados.
Se sigue en nuestro centro a 3 de los familiares portadores (III:2, III:3, III:21). Están clínicamente asintomáticos y sus exámenes cardiacos son normales. A diferencia de sus familiares y pese a estar asintomática, la paciente III:2 de 45 años presenta en el ENG signos de afección neuropática múltiple de intensidad moderada y signos compatibles con STC derecho.
A todos los portadores de la mutación se les ha ofrecido consejo genético y llevamos a cabo seguimiento a distancia de varios, con el fin de establecer el momento idóneo del TxH.
DiscusiónLa identificación de los pacientes cuya amiloidosis es de origen genético tiene gran importancia, ya que condiciona el tratamiento y tiene gran trascendencia para los familiares1, 2, 5.
Las causas hereditarias deben considerarse siempre en estos pacientes, ya que hasta el 10% de los sujetos diagnosticados de amiloidosis AL pueden padecer amiloidosis hereditarias5.
En nuestra experiencia, la utilización de técnicas inmunohistoquímicas facilita la identificación correcta del subtipo de amiloidosis en la biopsia (no necesariamente cardiaca) y evita diagnósticos erróneos y retrasos terapéuticos, como ocurrió en el primer paciente.
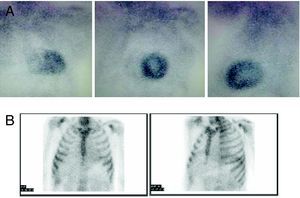
Para facilitar el diagnóstico del subtipo de amiloidosis cardiaca, se ha propuesto la gammagrafía 99mTc-DPD como una técnica que permite distinguir entre corazones con amiloide TTR (con captación) y amiloide AL (sin captación)6. Nuestra experiencia inicial con esta técnica en los 2 pacientes presentados y otros 4 con amiloidosis AL está en consonancia con lo publicado (Figura 3).

Figura 3. Gammagrafía 99mTc-DPD. A: amiloidosis hereditaria por transtiretina. B: amiloidosis AL. Obsérvese la diferente captación cardiaca. Cortesía del Dr. Javier de Haro del Moral.
La RM cardiaca permite una caracterización morfológica excelente, con la ventaja de permitir el estudio mediante realce tardío de gadolinio. El patrón de captación de gadolinio típico, se ha descrito como global y subendocárdico, pero también se ha descrito captación localizada o trasmural7. Desgraciadamente, a diferencia de la gammagrafía, la RM no permite diferenciar entre subtipos de amiloidosis.
Una vez establecido el diagnóstico de amiloidosis hereditaria e identificada la mutación, el estudio genético de los familiares permite establecer quiénes precisan de seguimiento y quiénes no. Como esto afecta a los descendientes de los no portadores, se reduce el número de familiares que hay que seguir.
Por otro lado, en los sujetos portadores, se puede realizar un consejo genético eficaz con implicaciones que van desde la planificación de descendencia a la carrera profesional. Así, por ejemplo, el sujeto III:3 (familia 2) trabaja como piloto comercial y, en su caso, el seguimiento estrecho de posible clínica neuropática condicionará su actividad laboral.
No recomendamos el estudio genético en menores de 16 años, debido a la nula actuación clínica derivada de conocer esta información y las posibles consecuencias psicológicas negativas del resultado para el menor y/o su entorno.
La atención a pacientes con amiloidosis hace necesario unidades multidisciplinarias con experiencia en complejos tratamientos y capacidad para proporcionar consejo genético3. En las amiloidosis hereditarias, el único procedimiento efectivo para tratar la fuente del amiloide es el TxH1, 2, 3, 8.
Aunque se ha descrito fenómeno de anticipación (aparición a edad más precoz en sucesivas generaciones) en formas neuropáticas con mutacion Val30Met, en las formas cardiacas es habitual que no aparezca afección clínica hasta la quinta década.
El seguimiento que aconsejamos para adultos portadores incluye revisión clínica y prueba de imagen cardiaca anual y ENG cada 2 años.
Dado que algún familiar evaluado ya tiene alteraciones neuropáticas preclínicas, se plantea cuándo debe realizarse el TxH que prevenga el desarrollo de afección cardiaca irreversible. La opinión de los expertos internacionales es que, dados los riesgos, el TxH debe realizarse en el primer año tras aparición de afección clínica8 (Rapezzi, comunicación personal).
Dado que el hígado de estos pacientes está por lo demás sano, puede ser empleado en un trasplante a receptores subóptimos (trasplante «dominó»). Desgraciadamente, el trasplante dominó puede ocasionar amiloidosis en el receptor.
Cuando hay afección cardiaca importante, la única alternativa es el trasplante combinado cardiohepático, tal como se planteó en nuestros pacientes.
Aunque se ha señalado que los depósitos cardiacos de amiloide mutado podrían actuar como «captadores» de proteína nativa9, creemos que no hay suficiente evidencia para sustentar en esto la necesidad de realizar trasplante simultáneo que, evidentemente, añade un riesgo significativo, y prácticamente el único beneficio que aporta es logístico.
En conclusión, la amiloidosis cardiaca hereditaria es una entidad de difícil diagnóstico y complejo tratamiento, cuya correcta identificación tiene importantes consecuencias en la actitud que adoptar con el enfermo y sus familiares.
Nuestra experiencia muestra la utilidad de efectuar estudio genético a familiares con el objetivo de realizar diagnóstico precoz, seguimiento estrecho y eventual tratamiento temprano.
FinanciaciónTrabajo parcialmente financiado por el FIS (PI08/979) y la red temática de investigación en insuficiencia cardiaca REDINSCOR (RD06/003/0018), Instituto de Investigación Carlos III, Ministerio de Sanidad, Política Social e Igualdad. Patricia Avellana recibe financiación como becaria de la Fundación Carolina-BBVA.
Conflicto de interesesNinguno.
Recibido 7 Junio 2010
Aceptado 12 Octubre 2010
Autor para correspondencia: Unidad de Miocardiopatías, Sección de Trasplante Cardiaco, Servicio de Cardiología, Hospital Universitario Puerta de Hierro. Manuel de Falla 2, 28222 Majadahonda, Madrid, España. pablogpavia@yahoo.es