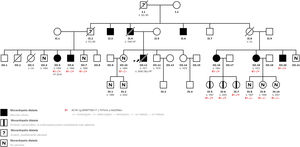

La miocardiopatía dilatada (MCD) es una enfermedad del miocardio caracterizada por dilatación y disfunción del ventrículo izquierdo (VI) o biventricular, no explicable por condiciones anómalas de carga o enfermedad coronaria, que causa insuficiencia cardiaca (IC), arritmias o muerte súbita (MS). Se ha demostrado una asociación familiar en un elevado porcentaje de los casos, lo cual resalta la importancia de los estudios genéticos de los familiares. Se presenta una familia con una mutación en el gen ACTA1 no descrita antes. Se obtuvo el consentimiento informado de todos los pacientes y la aprobación del comité de ética de investigación. En la figura 1 se muestra el árbol genealógico.

El caso índice es un varón de 36 años con asma bronquial como único antecedente de interés. Sus antecedentes familiares incluyen a un abuelo y un tío paternos con MS y su padre y otros 2 tíos paternos con MCD. Su presentación fue un primer episodio de IC en abril de 2005. Durante el ingreso, se diagnosticó al paciente de MCD y la ecocardiografía mostró una dilatación del VI con función sistólica global muy reducida e insuficiencia mitral grave. Se descartó enfermedad coronaria y, pese a la optimización del tratamiento, los síntomas persistieron. Se siguió al paciente en nuestra unidad de IC y cardiopatías familiares; se objetivó un deterioro progresivo de la fracción de eyección del VI (FEVI), y en julio de 2008 el paciente falleció tras un shock cardiogénico de rápida evolución.
En 2019, un varón de 55 años, primo del paciente índice, fue remitido a nuestra unidad (figura 1, caso III.7) para un cribado familiar. Su hermana (figura 1, caso III.5), que estaba en estudio en otro centro, presentó MCD en 2008, y se le implantó un desfibrilador automático implantable (DAI), que dio descargas apropiadas durante el seguimiento. En un panel de secuenciación masiva de 121 genes no se encontró ninguna mutación patogénica, pero sí se detectó una variante de significado incierto en el gen ACTA1. La paciente evolucionó rápidamente a IC avanzada, y precisó trasplante cardiaco en el mismo año del diagnóstico.
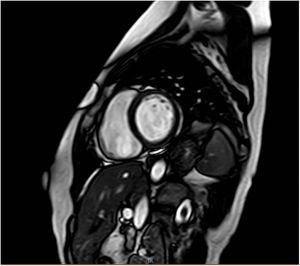
Durante el mismo periodo de tiempo, acudió a nuestro centro una mujer de 47 años (figura 1, caso III.16), prima del caso índice y de los casos III.5 y III.7. Las pruebas diagnósticas cardiacas mostraron una reducción leve de la FEVI (46%) sin dilatación ventricular, y la resonancia magnética mostró realce tardío de gadolinio lineal a nivel medio del segmento anteroseptal-septal-inferoseptal y basal medial (figura 2).

Se llevó a cabo un estudio cardiológico del caso III.7 en el que no se encontraron indicios de cardiopatía estructural. El estudio genético que se le hizo tanto a él como al caso III.16, mostró que ambos eran portadores de la variante heterocigota NM_001100,3:c.757G>A; p. (Gly253Ser) del gen ACTA1, que coincidía con la mutación descrita previamente en la paciente trasplantada. Durante el año siguiente, el cribado familiar se amplió para incluir a los familiares de primer grado, en los que el hallazgo más importante fue que 3 hijas del caso III.16 eran portadoras de la mutación del ACTA1 pero en ese momento no tenían una cardiopatía estructural asociada.
El gen ACTA1 codifica la isoforma de actina alfa, una proteína sarcomérica que participa en la contracción muscular y es especialmente abundante en los músculos esqueléticos. Las mutaciones de ACTA1 se han asociado clásicamente a diferentes tipos de miopatía del músculo esquelético. Aunque la afección cardiaca es infrecuente, algunos casos publicados han asociado el gen ACTA1 con cardiopatía en forma de MCD1 y miocardiopatía hipertrófica (MCH)2,3 acompañadas de miopatía. Llama la atención que la presentación predominante en la edad pediátrica sea la MCH y la MCD en adultos.
No obstante, hay muy pocos registros de mutaciones de este gen con afección exclusivamente miocárdica. El estudio de Carnevale A et al.4 en 55 pacientes con MCD a quienes se hizo un estudio genéticos amplio puso en evidencia que solo uno de los pacientes era portador de una mutación en ACTA1, aunque no se indicó si había miopatía. Hasta la fecha, tan solo hay una publicación que describa una mutación patogénica en ACTA1 con afección exclusivamente miocárdica5, concretamente la mutación heterocigota p.Arg256His, que se expresa en forma de MCD, arritmia ventricular y MS. Nuestro trabajo describe una nueva mutación en ACTA1, NM_001100,3:c.757G>A; p. (Gly253Ser), posiblemente asociada con MCD sin miopatía esquelética concomitante. Se trata de una variante tipo missense que afectaría a un aminoácido localizado en un dominio de la proteína relevante para el plegamiento de la actina. Además, la variante descrita por Reza et al.5 (p.Arg256His) se sitúa muy cerca de esta nueva variante (p.Gly253Ser) —a una distancia de solo 3 aminoácidos en la proteína—, lo cual lleva a plantear la hipótesis de que las variantes en esa región puedan estar relacionadas con este fenotipo y abre puertas a nuevas investigaciones. Hasta la fecha, no tenemos constancia de que esta variante se haya descrito asociada con el desarrollo de enfermedad ni consta en las bases de datos empleadas como control (ACTA1-LOVD variants database, ClinVar, Exome Variant Server, Exome Aggregation Consortium y Genome Aggregation Database).
Inicialmente se consideró de patogenicidad incierta pero, como se ha mostrado en el caso, al estudiar a gran parte de la familia, se observa cosegregación de esta variante con el fenotipo.
Este trabajo podría conducir a nuevos estudios, teniendo en cuenta la penetrancia, la progresión desfavorable y el muy mal pronóstico en los casos descritos, en los que el diagnóstico y el tratamiento precoces tienen especial interés. Deseamos resaltar también la importancia de las pruebas genéticas y los estudios familiares para verificar la cosegregación. A pesar de los avances en estos medios en los últimos años, no era fácil disponer de ellos en el momento de inicio de nuestro caso índice de la familia; si la disponibilidad hubiera sido más amplia, podría haberse tenido un diagnóstico más preciso, con una repercusión favorable en los demás familiares. Por último, como han resaltado también otros estudios, es necesario ampliar los conocimientos en este campo, pues resulta sorprendente que la actina alfa pueda producir una afección solo miocárdica, pese a que es una de las proteínas más ampliamente expresadas en el músculo esquelético.
FINANCIACIÓNNo se recibió financiación para este estudio.
CONTRIBUCIÓN DE LOS AUTORESA. Díaz Expósito: inclusión de los pacientes, exploraciones complementarias y estudio genético; revisión de la literatura; búsqueda de imágenes; versión inicial del artículo. A. Robles Mezcua: revisión de los estudios genéticos; elaboración del árbol genealógico familiar; versión inicial del artículo. I. Pérez Cabeza y J.M. García Pinilla: revisión de la carta científica.
CONFLICTO DE INTERESESNinguno de los autores tiene conflictos de intereses.