
El 15 de noviembre de 1992, hace ahora 30 años, el Journal of the American College of Cardiology publicó un artículo titulado «Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report»1. En él se describía a 8 pacientes con antecedente de muerte súbita (MS) por fibrilación ventricular (FV), reanimada. Después de una extensa investigación, no se encontró la causa de las arritmias en estos pacientes. Los 8 pacientes mostraron un electrocardiograma muy inusual con elevación del ST en las derivaciones precordiales derechas y lo que parecía ser un bloqueo de rama derecha. Las causas del síndrome se desconocían en ese momento, pero inmediatamente quedó claro que se trataba de un problema puramente eléctrico del corazón y que debía ser hereditario. La FV muy rápida indicaba un problema de dispersión de periodos refractarios cortos o normales, en contraste con la FV relativamente más lenta del síndrome de QT largo, en el que los periodos refractarios se prolongan debido a la repolarización prolongada1. Hoy se sabe que el patrón electrocardiográfico del síndrome de Brugada (SBr) es la vía final común de mecanismos fisiopatológicos diversos, algunos compartidos con alteraciones estructurales, aunque sin ellas en la mayoría de los casos.
Se habían tardado 5 años en recoger los datos de los primeros 4 pacientes, que se presentaron en un póster en la reunión de la North American Society of Pacing and Electrophysiology en 1991. Tras la presentación, y gracias a la colaboración internacional, se recopiló un gran número de posibles pacientes iguales. Finalmente, se seleccionó a 4 nuevos pacientes con idénticas características a los primeros. Esta colaboración internacional espontánea (sin financiación, sin protocolos, sin comités ni consejos) dio como resultado una de las publicaciones originales más citadas en cardiología. Lo que los autores consideraron inicialmente como una especie de curiosidad, luego se convirtió en una verdadera revolución científica (figura 1).
 desde su descripción hasta la actualidad.")
La descripción de este nuevo síndrome ha hecho mucha justicia al ECG como método de diagnóstico simple, económico pero muy valioso: el diagnóstico del SBr se basa en un ECG anormal. El llamado ECG «tipo 1» es la única condición para el diagnóstico, una vez descartadas otras posibles causas (fenocopias).
El SBr ha vuelto a dejar claro lo peligroso que es clasificar los ECG poco claros como variantes normales. El ECG del SBr fue considerado durante años una variante normal, sin significación diagnóstica ni pronóstica2. Tuvimos que darnos cuenta de nuestra ingenuidad de una manera muy dura ante la evidencia de que estos pacientes podían sufrir MS. Ha habido intentos de estructurar el diagnóstico de SBr a través de un sistema de puntos3. Por desgracia, esta puntuación no tiene ningún valor en la práctica, ya que hasta un 40% de los pacientes con SBr comprobado no tendrían los puntos suficientes para el diagnóstico4.
Definimos como patrón electrocardiográfico la presencia espontánea o en un test farmacológico de un patrón tipo 1. En caso de que haya síntomas (síncope, MS recuperada, fibrilación auricular o trastornos de la conducción), se habla de síndrome. La cuestión es si se puede hablar de enfermedad de Brugada en el momento en que se encuentra también una causa genética del síndrome.
FisiologíaEl SBr ha promovido el descubrimiento de nuevos mecanismos de arritmias: en particular, el fenómeno de la reentrada en fase 2 (R2P)5. El mecanismo correcto de la FV en el SBr aún está en debate. Además de la clásica reentrada basada en la conducción anormal, la R2P y la teoría de la cresta neural son dos alternativas para explicar las arritmias. El grupo de Ámsterdam considera la reentrada clásica en el tracto de salida del ventrículo derecho (TSVD) como el mecanismo más importante para la FV. Pero el grupo de Utica se apega a la teoría de la R2P. Mientras que en el primer mecanismo los potenciales de acción serían normales y el gradiente eléctrico se debería a la conducción lenta con potenciales de acción desfasados, en R2P el gradiente eléctrico está causado por un acortamiento de la duración del potencial de acción en el epicardio del TSVD. En el primer caso el problema reside en las mutaciones que reducen el flujo de sodio en la célula cardiaca; en el segundo, se basa en un flujo de potasio exagerado (corriente de iones de potasio). El grupo de Elizari propone que la base del SBr descansa en mutaciones en las células de la cresta neural que serían mutaciones somáticas. Para ellos, el SBr es un problema del desarrollo del corazón en la etapa embrionaria6. Esta posibilidad de mutaciones somáticas en el SBr también está respaldada por el hecho de que aproximadamente la mitad de los pacientes son casos aislados y no familiares, como si estos pacientes no pudieran transmitir la enfermedad a través de las células germinales. Es mejor decir que el SBr es solo un fenotipo con muchas posibles causas diferentes.
GenéticaLa descripción en 1998 del primer gen asociado al SBr marcó un verdadero punto histórico en las relaciones entre genética y cardiología. Hasta entonces, los estudios genéticos en cardiología eran muy escasos y los resultados se veían más como una curiosidad que como una contribución potencial para comprender los mecanismos y desarrollar un tratamiento. Pero entonces se pudo empezar a entender: el canal de sodio permanece abierto con ciertas mutaciones, la repolarización se prolonga y el paciente sufre QT largo. Si la corriente de sodio disminuye como resultado de otras mutaciones en el mismo gen, hay alteraciones de la conducción y SBr. De repente se abrió un mundo completamente nuevo.
No sorprende entonces que el número de mutaciones publicadas en todos los trastornos hereditarios cardiacos aumentara muy rápidamente. Con las nuevas técnicas para la investigación genética (estudio de asociación de genoma completo), todo el proceso de diagnóstico se ha acelerado exponencialmente. Toda esta nueva información viene con un problema de interpretación: ¿realmente importan todas las mutaciones y todos los genes?, ¿son la causa de la enfermedad?, ¿cuál es la importancia real de los polimorfismos? Desgraciadamente no tenemos los recursos, el tiempo y las suficientes cantidad y variedad de pacientes para estudiar funcionalmente cada mutación. Hay modelos para ayudarnos, pero los modelos siempre vienen con cierto grado de probabilidad e incertidumbre.
FertilidadCon todas las limitaciones que uno pueda imaginar, el diagnóstico genético preimplante (DGP) se ha convertido en una opción obvia para tratar enfermedades hereditarias. Quienes se oponen a la técnica argumentan que casi ninguna enfermedad, especialmente el SBr, es monogenética. Además del gen principal considerado causa de la enfermedad, debe haber otras mutaciones y variaciones, incluidos los polimorfismos, que se acumulan hasta que se alcanza cierta puntuación de riesgo genético. Por lo tanto, implantar un embrión seleccionado en función de la ausencia de una mutación en el canal de sodio no tendría ningún valor para prevenir el SBr. Los defensores del DGP dicen, con los mismos argumentos, que simplemente seleccionar un embrión sin la mutación baja esa puntuación de riesgo genético, y así se puede contrarrestar la manifestación de la enfermedad. El DGP se ofrece en nuestro hospital desde hace años para más de 200 enfermedades diferentes que se consideran monogenéticas, incluido el SBr7. Si se identifica una mutación de clase 4 o 5, nuestra práctica habitual es ofrecer un DGP, con lo que se trata de evitar la transmisión de la enfermedad.
PediatríaEl SBr es una causa de MS en los niños y también una de varias posibles causas del síndrome de MS del lactante. En pocas enfermedades se ha especulado durante tanto tiempo y de forma tan esotérica como en el caso de la MS del lactante. Ahora sabemos que la mayoría de estas muertes súbitas se deben a arritmias, incluido el SBr8.
Nos encontramos con un problema similar en el diagnóstico de epilepsia y síncope de causa desconocida en niños. No solo el QT largo, sino también el síndrome de QT corto y el SBr deben incluirse en el diagnóstico diferencial, más aún en casos difíciles de tratar. Tampoco hay que olvidar que los pacientes también pueden padecer más de una enfermedad: epilepsia y SBr conjuntamente9 y también síncope vasovagal y arrítmico simultáneamente.
La estrategia diagnóstico-terapéutica del SBr en edad pediátrica es especialmente delicado y la evidencia científica es escasa. Nos encontramos con dos problemas: la estrategia en niños con diagnóstico de SBr y el estudio de los hijos de los pacientes. En el primer caso nuestra actitud es similar que con los adultos, con un seguimiento estrecho e implante de un desfibrilador automático implantable (DAI) en caso presencia de factores de alto riesgo. En el caso de hijos de pacientes, realizamos un test farmacológico a los 12 y los 18 años de edad y en caso de que se presenten síntomas. Hasta un 25% de los niños con un test negativo a la edad de 12 años son positivos a los 18 y los síntomas pueden preceder a la positividad del test o la aparición de un patrón tipo 1 espontáneo10. Prestamos especial atención a la presencia de disfunción sinusal, ya que en nuestra experiencia se asocia con un muy mal pronóstico y a estos niños se les debe implantar un DAI.
Medicina deportivaNada afecta más a nuestra imaginación que la muerte repentina de un atleta «perfectamente sano». No todas las MS de deportistas ocurren durante el ejercicio. En realidad, es lo opuesto10. La mayoría muere de repente después del esfuerzo, inmediatamente o más tarde, en completo reposo. Se sabe desde hace mucho tiempo que el síndrome de QT largo y la taquicardia ventricular polimórfica dependiente de catecolaminas eran una causa. Pero ahora, después de un examen detallado de los familiares de los fallecidos, parece que la causa más frecuente es el SBr11.
Consideramos adecuada la realización de al menos un ECG a las personas que practiquen deporte de modo habitual. La hipertermia durante la práctica deportiva o estados vagotónicos al cesar el esfuerzo pueden inducir un patrón tipo 1 o desencadenar un síncope o MS en pacientes con SBr de alto riesgo. En caso de síntomas, antecedentes familiares de MS a edad precoz o un ECG con un patrón tipo 2, proponemos la realización de un test de provocación farmacológica.
Medicina medicolegalLos resultados de las autopsias varían enormemente de un estudio a otro y con la experiencia de los médicos y su empeño en buscar una causa. Aquí entra en juego el estudio de los familiares y la «autopsia molecular». El estudio de Papadakis et al.11 mostró que la causa más frecuente de MS, cuando se encuentra la causa, es el SBr. Esto se demostró a través de los resultados de la prueba de ajmalina en familiares. Las pruebas genéticas post mortem también pueden revelar una posible mutación causal en el 20-40% de los casos12.
Medicina preventivaExaminar a las personas que parecen saludables es una de las mejores formas de descubrir enfermedades ocultas. Pero, por supuesto, el valor de la detección está muy relacionado con el investigador y las pruebas realizadas.
En relación con las enfermedades cardiacas hay gran controversia: a favor del cribado, los estudios italianos13, o en contra, los estadounidenses. Hay, en todo caso, una gran diferencia en los argumentos. Mientras que los italianos se basan en una reducción de la incidencia de MS gracias al cribado, los argumentos estadounidenses en contra son puramente económicos. Pero ¿qué precio se pone a la vida de un joven?
Dispositivos electrónicos cardiacos y ablación del TSVDEl SBr es una enfermedad de jóvenes. Solo se puede controlar (en términos de MS) mediante el implante de un DAI. No es de extrañar, entonces, que las técnicas de implante se hayan adaptado a los niños. Por ejemplo, un implante abdominal subcostal es mucho más cómodo que uno prepectoral, sobre todo en lo que se refiere a la práctica deportiva. El implante se puede hacer con electrodos epicárdicos, de modo que el sistema venoso del paciente queda completamente intacto. En centros con experiencia, este implante puede combinarse con una ablación epicárdica del TSVD, donde se encuentra el sustrato del SBr14. Esta combinación es nuestro protocolo actual para el tratamiento del SBr. Debe quedar claro que aún no hay suficientes datos sobre los efectos a largo plazo de la ablación. Actualmente, la ablación no es una alternativa al DAI.
Aspectos del sexoEn nuestra experiencia, la idea de que el SBr afecta menos a las mujeres es falsa. Prácticamente el 50% de nuestros pacientes son mujeres15. El hecho de que muestren menos síntomas puede llevar a que se diagnostique menos a las mujeres, pero con un meticuloso cribado de familiares y una alta sospecha clínica la tasa de diagnóstico de las mujeres es similar a la de los varones.
Muchas publicaciones sobre SBr siempre han enfatizado que los varones tienen peor pronóstico que las mujeres. Si bien esta idea parece ser cierta en adultos, no lo es antes de la pubertad. No se han encontrado diferencias en los síntomas y la mortalidad entre niños y niñas prepúberes10. Está muy claro que la testosterona tiene parte en el SBr. Se ha demostrado que la castración masculina mejora las manifestaciones de la enfermedad16.
Estratificación del riesgoUn aspecto fundamental del SBr es la evaluación del riesgo de MS. El SBr tiene una presentación clínica muy amplia. El diagnóstico se puede realizar tras una MS recuperada, pero cada vez son más los pacientes completamente asintomáticos. Síncope, fibrilación auricular, disfunción sinusal y alteraciones de la conducción son síntomas y hallazgos con impacto en el pronóstico. Pero la pregunta es: ¿a quién debe implantarse un DAI de forma preventiva? La mitad de los pacientes que sufrieron MS no tenían síntomas previos.
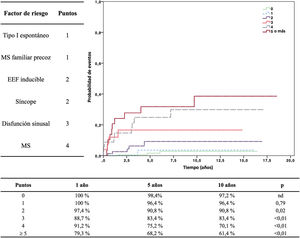
En un intento de integrar la información disponible, hemos desarrollado un sistema de puntuación para establecer el riesgo de eventos arrítmicos17 (figura 2). Este sistema de estratificación de riesgo es muy valioso, pero conlleva una gran paradoja: se puede cometer el error de asumir un riesgo bajo como si significara ningún riesgo. A los pacientes con una puntuación baja, por lo tanto, no se los considera candidatos para la protección con DAI. Por el contrario, se protege sistemáticamente a los pacientes con una puntuación alta. El resultado, paradójicamente, es que los pacientes con alto riesgo sobreviven a los eventos arrítmicos gracias a la protección del DAI, mientras que los del grupo con bajo riesgo, si se presentara una arritmia, morirían por falta de protección con un DAI. Así, aunque la incidencia de arritmias es mucho menor en la categoría de bajo riesgo, la verdadera mortalidad es la más alta por falta de protección.

Estratificación del riesgo del paciente con síndrome de Brugada. A la izquierda se muestran los factores de riesgo con su valor en puntos. La gráfica muestra los eventos arrítmicos según la puntuación. Reproducida con permiso de Sieira et al.17. EFF: estudio electrofisiológico; MS: muerte súbita.
En 30 años hemos aprendido mucho sobre el SBr, pero también sobre otras enfermedades relacionadas. Está claro que con la descripción del SBr todo el mundo de la arritmología ha entrado en una nueva dimensión.
FINANCIACIÓNNo se ha recibido financiación.
CONTRIBUCIÓN DE LOS AUTORESLos tres autores han contribuido por igual a la redacción y la revisión del artículo.
CONFLICTO DE INTERESESP. Brugada ha recibido compensación con fines docentes de Biotronik. C. de Asmundis recibe subvenciones de investigación a nombre de su centro de Biotronik, Medtronic, Abbott, Boston Scientific, AtriCure, Philips y Acutus y ha recibido compensación con fines docentes y de proctoring de Medtronic, Abbott, Biotronik, Boston Scientific, Atricure, Acutus Medical y Daiichi Sankyo. J. Siera no tiene conflicto de intereses.