
La resonancia magnética se ha convertido en técnica de imagen de primera línea en muchas situaciones clínicas. El número de pacientes portadores de dispositivos cardiovasculares, como los dispositivos cardiovasculares electrónicos implantables, ha crecido de modo exponencial. Aunque se han descrito complicaciones y efectos adversos cuando estos pacientes se someten a exploraciones de resonancia magnética, la evidencia clínica actual respalda la seguridad de realizar estos estudios cuando se cumplen unas normas y recomendaciones dirigidas a minimizar los posibles riesgos. El Grupo de Trabajo de Cardiorresonancia Magnética y Cardiotomografía Computarizadas de la Sociedad Española de Cardiología (SEC-GT CRMTC), la Asociación del Ritmo Cardiaco de la Sociedad Española de Cardiología (SEC-Asociación del Ritmo Cardiaco de la Sociedad Española de Cardiología), la Sociedad Española de Radiología Médica (SERAM) y la Sociedad Española de Imagen Cardiotorácica (SEICAT) han elaborado el presente documento, que revisa la evidencia disponible en este campo y establece las recomendaciones necesarias para que los pacientes portadores de dispositivos cardiovasculares electrónicos implantables y otros dispositivos puedan acceder con seguridad a este instrumento diagnóstico.
Palabras clave
En los últimos años, la resonancia magnética (RM) se ha convertido en una técnica de imagen esencial y de primera línea en muchas situaciones clínicas. Al mismo tiempo, el número de pacientes con diferentes dispositivos cardiovasculares ha crecido de manera exponencial. Por lo tanto, es habitual en la práctica clínica encontrarnos con pacientes portadores de alguno de estos dispositivos que precisen de un estudio de RM1,2. Se estima que la probabilidad de requerir un estudio de RM en el primer año tras recibir un dispositivo cardiaco electrónico implantable (DCEI) es del 10% y a lo largo de la vida del paciente, del 75%3.
Aunque en muchos casos estos dispositivos no suponen una contraindicación para la RM, en otros los dispositivos no son seguros, son compatibles solo en determinadas circunstancias o requieren una valoración antes y después de la prueba. Además, dado que este tipo de materiales puede condicionar una menor calidad de imagen, la indicación de la prueba debe valorarse en el contexto de su riesgo/beneficio.
El presente documento de consenso se ha elaborado mediante la colaboración entre el Grupo de Trabajo de Cardiorresonancia Magnética y Cardiotomografía Computarizadas de la Sociedad Española de Cardiología (SEC-GT CRMTC), la Asociación del Ritmo Cardiaco de la SEC (SEC-Asociación del Ritmo Cardiaco), la Sociedad Española de Radiología Médica (SERAM) y la Sociedad Española de Imagen Cardiotorácica (SEICAT). En él se pretende hacer una revisión de la seguridad de los estudios de RM de pacientes portadores de DCEI y otros dispositivos cardiovasculares y establecer unas recomendaciones prácticas para que todos los pacientes portadores puedan acceder con seguridad a este tipo de exploraciones diagnósticas. En el anexo 1 y el anexo 2 se puede consultar la filiación de cada autor y revisor del documento a las entidades impulsoras del presente documento.
DEFINICIONESLa RM es una técnica que permite obtener imágenes gracias a la interacción entre los campos magnéticos y los núcleos de hidrógeno de los diferentes tejidos. Para generar las imágenes, es necesaria la aplicación de 3 tipos de campos magnéticos que tienen un efecto diferente en el cuerpo humano y los objetos circundantes y afectan a la seguridad de las exploraciones4,5.
- 1.
El campo magnético estático (B0). Es el campo magnético del equipo, siempre está encendido y su intensidad se mide en teslas (T); los equipos más empleados los de 1,5 y 3 T. La interacción con elementos ferromagnéticos puede hace que los objetos se muevan, se desplacen o se aceleren hacia el imán. Sin embargo, este riesgo generalmente no se aplica a dispositivos médicos implantados por su composición mayoritaria de materiales no ferromagnéticos.
- 2.
Gradientes dinámicos que causan cambios en el campo magnético a lo largo del tiempo (dB/dt). Se trata de gradientes que se activan y desactivan rápidamente durante los estudios de RM. Estos gradientes pueden inducir corrientes eléctricas en determinados dispositivos en forma de calentamiento, vibración, estimulación neuromuscular y ruido acústico.
- 3.
Campos magnéticos de radiofrecuencia (RF) (B1). Se producen fundamentalmente en exploraciones del área torácica, región de la antena que emite la RF. Parte de la energía aplicada es absorbida por el cuerpo (medida por la tasa de absorción específica en W/kg) y convertida en calor, su principal efecto biológico. La tasa de absorción específica aumenta con el campo magnético y varía con las diferentes secuencias. Los campos magnéticos de RF presentan riesgo de interferencia electromagnética con los DCEI.
Según el perfil de seguridad cuando se los somete a exploraciones de RM, los dispositivos cardiovasculares se clasifican de la siguiente forma:
- 1.
Dispositivo compatible con RM. Aquel que siempre puede someterse a una RM con seguridad.
- 2.
Dispositivo condicional. Dispositivos seguros en un entorno de RM teniendo en cuenta una serie de consideraciones. En el caso de DCEI, son dispositivos modificados en su hardware (minimización del material ferromagnético y modificación de electrodos) y software que permiten realizar con seguridad un estudio de RM en determinadas condiciones6.
- 3.
Dispositivo no condicional. Dispositivos con los que, por su diseño o funcionamiento, no se pueden asegurar las condiciones óptimas de seguridad en la realización del estudio de RM. En el caso de DCEI, se ha demostrado que estos dispositivos pueden someterse a RM con baja incidencia de complicaciones siempre que se tomen determinadas precauciones en la programación del dispositivo, la monitorización del paciente y las características de la RM7,8.
Se debe tener en cuenta que para determinar la compatibilidad de un dispositivo o conjunto de dispositivos (sistema), todos sus componentes deben ser compatibles o condicionales con RM. En el caso de los DCEI, se precisa que tanto los componentes (generador y electrodo) como su combinación (idealmente misma marca y combinación validada en entorno de RM) deben ser compatibles y, en caso preciso, se deben cumplir condiciones de campo magnético y tasa de absorción específica, determinadas para cada modelo9.
Aunque los estudios de RM se pueden realizar a pacientes con dispositivos compatibles o condicionales con seguridad, la presencia de estos puede afectar a la calidad de la imagen y, por lo tanto, a la rentabilidad de la prueba. Esto dependerá del tipo de dispositivo, su localización, el área anatómica en estudio y el tipo de secuencias empleadas. Este factor se debe tener en cuenta en la valoración previa del paciente cuando se plantee un estudio de RM.
INTERACCIÓN ENTRE RM Y DISPOSITIVOS CARDIOVASCULARES. SEGURIDADInfluencia de los dispositivos en la imagen de RM (artefactos)Los artefactos en la RM son relativamente frecuentes. Se definen como cualquier aumento o pérdida de señal que no tiene base anatómica o es la consecuencia de la distorsión, adición o perdida de información, en general relacionados con la existencia de materiales metálicos o ferromagnéticos. Aunque algunos son obvios y fácilmente reconocibles, otros son mucho más sutiles y pueden llevar a errores de interpretación y diagnóstico.
En general, hay 2 tipos de artefactos inducidos por materiales ferromagnéticos:
- 1.
Artefactos de susceptibilidad magnética. Son la principal causa de aparición de los artefactos y se deben a la falta de homogeneidad local del campo producidas por la presencia de materiales ferromagnéticos dentro del imán.
- 2.
Artefactos por corrientes de Foucault (también llamado de corrientes de Eddy). Ocurren porque los gradientes de los pulsos de RF inducen corrientes eléctricas en los materiales metálicos circundantes, crean campos magnéticos intermitentes no deseados que degradan la homogeneidad del campo y distorsionan la imagen.
Por lo general, el artefacto aparece como bandas con aumento o disminución de la intensidad de la señal alrededor de las partes metálicas. Puesto que deterioran la imagen de resonancia, deben reconocerse, ya que se pueden confundir con una imagen patológica10. La magnitud de los artefactos dependerá tanto de las características intrínsecas del dispositivo (composición metálica, tamaño y orientación con respecto a la dirección del campo magnético) como de la distancia a la que se localice de la región anatómica en estudio (poco frecuentes en estudios fuera del área torácica), la secuencia utilizada y la intensidad del campo magnético.
Influencia de la RM en los dispositivosEn términos generales, los dispositivos cardiovasculares con mayor influencia/interferencia generada por el estudio RM son los dispositivos de asistencia y monitorización circulatoria (dispositivos de asistencia ventricular izquierda, sistemas de oxigenación extracorpórea de membrana, catéter de gasto continuo) y los DCEI (marcapasos, DAI, dispositivos de terapia de resincronización cardiaca y electrodos abandonados). Los primeros suponen una contraindicación absoluta. En el caso de los DCEI, las complicaciones son poco frecuentes, pero potencialmente graves2,11. Por su mayor frecuencia en el contexto clínico, en la tabla 1 se detallan los potenciales efectos del entorno de RM en los DCEI.
Potenciales efectos del entorno de la resonancia magnética en los dispositivos cardiovasculares electrónicos implantables
| Efectos de la RM en DCEI | Explicación |
|---|---|
| Calentamiento y lesión del tejido circundante | Los electrodos pueden actuar como antenas de la energía electromagnética y generar corrientes dentro del sistema que aumentarían la temperatura del tejido circundante, que podrían dañar el miocardio local, aumentar el umbral de captura, reducir la amplitud de la onda sensada o, teóricamente, aumentar el umbral de desfibrilación de los DAIEn estudios in vitro, el aumento de temperatura es mayor en los electrodos dañados, abandonados (ya que no existe conexión a un generador que actúe como disipador del calor) y/o epicárdicos (dada la ausencia de enfriamiento convectivo por ser un espacio sin flujo sanguíneo). La situación es potencialmente peor en los electrodos epicárdicos abandonados |
| Desplazamiento | El campo magnético podría producir desplazamientos del material ferromagnético del generador, pero es algo realmente excepcional. Los dispositivos compatibles con RM reducen este riesgo por su menor cantidad de material ferromagnéticoLa mayoría de los fabricantes aconsejan esperar 6 semanas tras el implante (proceso de «fijación» del electrodo por cicatrización), si bien existen series de pacientes sometidos a RM en los primeros días tras el implante sin complicaciones, por lo que la RM podría ser precoz si fuese clínicamente necesario12 |
| Estimulación asíncrona | Los pulsos de RF de la RM atraviesan el electrodo y pueden estimular el tejido e inducir arritmias auriculares y/o ventriculares |
| Sobresensado | Los pulsos de RF pueden generar «ruido» (sobresensado) que dé lugar a la inhibición del impulso del marcapasos (riesgo de asistolia en pacientes dependientes) o terapias antitaquicardia inapropiadas del DAI (riesgo de terapias inapropiadas) |
| Interruptor de láminas activado magnéticamente | Al acercar un imán al dispositivo, normalmente el dispositivo estimula en modo asíncrono e inhibe las terapias antitaquicardia del DAI. Este comportamiento se modifica en los DCEI compatibles cuando el modo de compatibilidad se encuentra activado |
| Reseteo eléctrico | La interferencia electromagnética generada por la RM puede generar el reseteo eléctrico de los DCEI pasando a una programación que es específica de cada fabricante y modelo. Generalmente activa el modo VVI (riesgo de asistolia en pacientes dependientes) y se activan las terapias antitaquicardia según parámetros de diagnóstico y terapia nominales, habitualmente en una única zona de fibrilación ventricular con descargas de alta energía (riesgo terapias inapropiadas) |
| Drenaje anómalo de la batería | Excepcionalmente podría producirse agotamiento de la batería, más frecuente en DCEI próximos al recambio electivo del generador (batería en límite bajo)13. Es una complicación importante que conlleva el recambio del generador del DCEI |
DAI: desfibrilador automático implantable; DCEI: dispositivo cardiovascular electrónico implantable; RF: radiofrecuencia; RM: resonancia magnética.
En los últimos años, son numerosos los trabajos que han demostrado la seguridad de las exploraciones de RM cardiotorácica y no cardiotorácica en pacientes portadores de DCEI siempre que se adopte una serie de medidas apropiadas2,11.
Los trabajos publicados hasta la fecha son bastante homogéneos en su diseño (descripción de los posibles efectos adversos, programación del dispositivo, secuencias realizadas) y los resultados, concordantes y no se documentan complicaciones clínicamente significativas derivadas de la realización de los estudios en imanes de 1,5 y 3 T siguiendo unas pautas de seguridad definidas14,15. En la tabla 2 se presenta un resumen de los principales trabajos en este campo.
Principales estudios clínicos multicéntricos y metanálisis que han evaluado la seguridad de los estudios de resonancia magnética para los pacientes portadores de distintos tipos de dispositivos cardiovasculares electrónicos implantables
| Tipo de estudio | Tipo de dispositivo | Campo/tipo de RM/región anatómica | Pacientes | Complicaciones de RM | Cambio en los parámetros del dispositivo | Referencia |
|---|---|---|---|---|---|---|
| Estudios multicéntricos | Dispositivo condicional (9-12 semanas tras el implante) | 1,5 TSAR≤2 W/kgCerebral/lumbar | 464 | Ninguna | Mínimos | Wilkoff et al.16, 2011 |
| Dispositivo condicional (9-12 semanas tras el implante) | 1,5 TSAR≤2 W/kgCualquiera | 263 | Ninguna | — | Gimbel et al.17, 2013 | |
| DAI condicional | 1,5 TSAR≤2 W/kgTorácica, cuello, cerebral | 275 | Ninguna | Mínimos | Gold et al.18, 2015 | |
| Dispositivo condicional | 1,5 TSAR≤2 W/kgCardiaca y columna dorsal | 245 | 1 efecto adverso | Sin cambios en estimulación y mínimos en sensado | Bailey et al.19, 2015 | |
| Dispositivo condicional | 1,5 TSAR≤2 W/kgCerebral y lumbar baja | 226 | Ninguna | Mínimos | Bailey et al.20, 2015 | |
| DAI condicional | 1,5 TSAR≤2 W/kgColumna dorsal y cardiaca | 170 | Ninguna | Ninguno | Awad et al.21, 2015 | |
| Dispositivo condicional | 1,5 TSAR≤2 W/kgTorácica y cerebral | 266 | Ninguna | Mínimos en estimulaciónNinguno en sensado | Shenthar et al.22, 2015 | |
| Dispositivo condicional | 1,5 TSAR≤2 W/kgSlew rate≤200 T/m/sCualquier región anatómica | 526 | 2 FA paroxística2 calentamientos1 fallo de captura1 incremento del umbral | 4 pacientes (0,76%) con umbral> 0,5 V e incremento en umbrales | Williamson et al.23, 2017 | |
| Dispositivos no condicionales (DAI dependientes excluidos) | 1,5 TRM no torácica | 1.500 | 5 FA y 1 aleteo auricular6 reseteos eléctricos parciales | Ligero incremento umbral estimulaciónLigera disminución de la sensibilidad | Russo et al.24, 2017 | |
| Marcapasos sin cables | 1,5 T/3 T | 14 | Ninguna | Micra (Medtronic, Estados Unidos), ninguno | Blessberger et al.25, 2019 | |
| Metanálisis | Dispositivos no condicionales | 1,5, 2/3 TCualquiera | 5.099 | 3 fallos del electrodo94 reseteos (ninguno en posteriores a 2006)11 estimulación no apropiada17 síntomas | Sin cambios clínicamente significativos | Shah et al.26, 2018 |
| Dispositivos no condicionales | 1,5/3 TCualquiera | 5.625 | 1 fallo del electrodo2 fallos del generador76 reseteos encendido-apagado6 estimulaciones inapropiadas19 síntomas | Incremento> 0,5 V en el umbral de estimulación y cambios> 50 Ω en la impedancia (el 1,1 y el 4,8%). Cambios significativos en la onda P y onda R (el 1,5 y el 0,4%) | Glikson et al.7, 2020 |
DAI: desfibrilador automático implantable; FA: fibrilación auricular; RM: resonancia magnética; SAR: tasa de absorción específica.
Los cables epicárdicos y los electrodos abandonados tienen un mayor riesgo de calentamiento durante las exploraciones de RM. La evidencia disponible en este campo es muy escasa (inexistente en el caso de los cables epicárdicos) y por ello no se recomienda la RM para estos pacientes y únicamente podría valorarse para casos de pacientes en una situación clínica grave sin otra alternativa diagnóstica. No obstante, un trabajo de reciente publicación, con 139 pacientes y 243 electrodos abandonados, respalda la seguridad de la RM en esta situación clínica8. Sin embargo, en los marcapasos temporales, las características del generador y electrodo presentan mayor de riesgo de complicaciones, por lo que no se recomienda someterlos a este tipo de exploraciones en ninguna circunstancia, aunque no se disponga de evidencia científica al respecto12.
En cuanto a los dispositivos de estimulación sin cables, aunque la experiencia es menor, los trabajos iniciales no demuestran una mayor tasa de complicaciones y parecen seguros tanto en 1,5 como en 3 T. Los DAI subcutáneos no requieren un manejo diferente que el de los DAI convencionales, y se debe verificar, en función del modelo y el año de fabricación, si son condicionales o no condicionales para los estudios de RM12.
Los dispositivos de grabación en bucle se consideran seguros y no suponen nunca una contraindicación para las exploraciones27.
Otros dispositivos cardiovascularesEn los últimos años, el número y los tipos de dispositivos cardiovasculares o vasculares que se implantan para el tratamiento de distintas afecciones está creciendo de manera exponencial. Aunque gran parte de ellos son seguros o condicionales en el entorno de la RM, se recomienda comprobar su compatibilidad especifica. En la tabla 3 se recogen los utilizados más habitualmente. A modo de resumen, se pueden considerar seguros, respetando algunos parámetros en la adquisición, los stents coronarios y vasculares, los tubos vasculares y los parches quirúrgicos, las prótesis valvulares quirúrgicas (biológicas y mecánicas), las prótesis y los dispositivos transcatéter (oclusores septales, cierre de orejuela, reemplazo y reparación valvular) y las grabadoras de eventos en bucle.
Principales tipos de dispositivos sin estimulación utilizados actualmente en cardiología y cirugía cardiaca, compatibilidad y seguridad con resonancia magnética en equipos de 1,5 y 3 T
| Grupo del dispositivo | Tipo de dispositivo | Realización de estudio de RM | Observaciones | Referencia | |
|---|---|---|---|---|---|
| 1,5 T | 3 T | ||||
| Vascular y coronario | Stent coronario | Seguro | Seguro | Existe numerosa literatura en infarto agudo miocardio (desde 1 h a 7 días tras ICP) sin complicaciones comunicadas | Shellock et al.4Symons et al.5, 2019Levine et al.28, 2007Jehl et al.29, 2009Patel et al.30, 2006Karamitsos et al.31, 2017Kaya et al.32, 2009Curtis et al.33, 2013 |
| Tubos vasculares | Seguro | Seguro | - | Symons et al.5, 2019Jehl et al.29, 2009Karamitsos et al.31, 2017Curtis et al.34, 2006 | |
| Endoprótesis aórticas | Condicional (seguir indicaciones del fabricante) | Condicional (seguir indicaciones del fabricante) | GEM: 720 Gauss/cmSAR: 3 W/kg por 15 min de examen | Symons et al.5, 2019Levine et al.28, 2007Jehl et al.29, 2009Dill et al.35, 2008 | |
| Stents aórticos no recubiertos | Condicional (seguir indicaciones del fabricante) | Condicional (seguir indicaciones del fabricante) | GEM:720 Gauss/cmSAR: 2 W/kg por 15 min de examen | Symons et al.5, 2019Levine et al.28, 2007Jehl et al.29, 2009Dill et al.35, 2008Grzyska et al.36, 2021 | |
| Valvular quirúrgico | Prótesis valvulares biológicas | Seguro | Condicional (seguir indicaciones del fabricante) | - | Shellock et al.4Symons et al.5, 2019Dill et al.35, 2008Myers et al.37, 2012Baikoussis et al.38, 2011 |
| Prótesis valvulares mecánicas | Seguro | Condicional (seguir indicaciones del fabricante) | - | Symons et al.5, 2019Levine et al.28, 2007Dill et al.35,2008Myers et al.37, 2012 | |
| Conductos valvulados mecánicos | Seguro | Condicional (seguir indicaciones del fabricante) | - | Symons et al.5, 2019Levine et al.28, 2007Karamitsos et al.31, 2017Dill et al.35, 2008Myers et al.37, 2012Baikoussis et al.38, 2011 | |
| Conductos valvulados biológicos | Seguro | Condicional (seguir indicaciones del fabricante) | - | Symons et al.5, 2019Levine et al.28, 2007Karamitsos et al.31, 2017Dill et al.35, 2008Myers et al.37, 2012Baikoussis et al.38, 2011 | |
| Homoinjertos/xenoinjertos | Seguro | Seguro | — | Symons et al.5, 2019Levine et al.28, 2007Karamitsos et al.31, 2017Dill et al.35, 2008Myers et al.37, 2012Baikoussis et al.38, 2011 | |
| Anuloplastias quirúrgicas | Seguro | Seguro | Algunos modelos especifican indicaciones para escanear inmediatamente tras el implante | Shellock et al.4Symons et al.5, 2019Myers et al.37, 2012 | |
| Valvular percutáneo | Prótesis valvular transcatéter | Seguro | Seguro | TAVI (CoreValve, Medtronic, Estados Unidos; SAPIEN, Edwards Lifesciences, Estados Unidos): se consideran seguros en sistemas de 1,5 y 3 T. Se puede escanear inmediatamente tras la colocación siguiendo las indicaciones (GEM: 720 Gauss/cm; SAR: 2 W/kg por 15 min de examen). Aplica para TAVI en otras posiciones valvulares (pulmonar, mitral o tricúspide) | Shellock et al.4Shellock et al.39, 2001Saeedi et al.40, 2015Hartlage et al.41, 2016 |
| Seguro | Seguro | TMVR (Tendyne, Abbott, Estados Unidos) | Lin et al.42, 2018 | ||
| Reparación valvular transcatéter | Seguro | Seguro | Reparación borde-borde (Mitraclip, Abbott, Estados Unidos) | Shellock et al.4Lurz et al.43, 2011 | |
| Condicional (seguir indicaciones del fabricante) | Condicional (seguir indicaciones del fabricante) | Anuloplastia directa (Cardioband, Edwards Lifesciences, Estados Unidos): se puede escanear inmediatamente tras la colocación siguiendo las indicaciones (GEM: 720 Gauss/cm; SAR: 2 W/kg por 15 min de examen) | Shellock et al.4Symons et al.5, 2019 | ||
| Percutáneo no valvular | Oclusores septales (FOP, CIA, CIV) | Condicional (no si se sospecha pérdida de integridad) | Condicional (no si se sospecha pérdida de integridad) | Puede considerarse un periodo ventana de 6 semanas tras el implante. Si se sospecha pérdida de integridad del dispositivo, se desaconseja | Shellock et al.4Myers et al.37,2012Shellock et al.44, 2005 |
| Cierre de orejuela | Condicional (no si se sospecha pérdida de integridad) | Condicional (no si se sospecha pérdida de integridad) | Puede considerarse un periodo ventana de 6 semanas tras el implante. Si se sospecha pérdida de integridad del dispositivo, se desaconseja | Shellock et al.4Myers et al.37, 2012Mohrs et al.45, 2011 | |
| Otros cierres (PVL, DAP) | Condicional (no si se sospecha pérdida de integridad) | Condicional (no si se sospecha pérdida de integridad) | Puede considerarse un periodo ventana de 6 semanas tras el implante. Si se sospecha pérdida de integridad del dispositivo, se desaconseja | Shellock et al.4Symons et al.5, 2019Myers et al.37, 2012 | |
| Monitorización y asistencia circulatoria | Asistencias ventriculares (BCPIAo, ECMO, LV/RVAD) | No seguro | No seguro | — | Symons et al.5, 2019Levine et al.28, 2007Dill et al.35, 2008Baikoussis et al.38, 2011Lee et a.46, 2014 |
| Catéter Swan-Ganz | No seguro | No seguro | Se podría realizar si el catéter no tiene sistemas de termodilución o electrodos de estimulación | Shellock et al.4Symons et al.5, 2019Dill et al.35,2008 | |
| Catéter gasto continuo | No seguro | No seguro | Symons et al.5, 2019Levine et al.28, 2007Dill et al.35,2008 | ||
| Monitor continuo de la presión pulmonar | Seguir indicaciones del fabricante | Seguir indicaciones del fabricante | CardioMEMS, Abbott, Estados Unidos: se puede escanear siguiendo las indicaciones (GEM: 720 Gauss/cm. SAR: 2 W/kg por 15 min de examen) | Shellock et al.4Symons et al.5, 2019 | |
| Otros | Grabadores de eventos | Seguir indicaciones del fabricante | Seguir indicaciones del fabricante | GEM: 720 Gauss/cm | Symons et al.5, 2019Levine et al.28, 2007Baikoussis et al.38, 2011 |
| Parches quirúrgicos | Seguro | Seguro | - | Symons et al.5, 2019Levine et al.28, 2007Dill et al.35, 2008Myers et al.37, 2012 | |
| Cerclajes metálicos esternales | Seguro | Seguro | - | Shellock et al.4Symons et al.5, 2019 | |
BCPIAo: balón de contrapulsación intraaórtico; CIA: comunicación interauricular; CIV: comunicación interventricular; DAP: ductus arterioso persistente; ECMO: oxigenador extracorpóreo de membrana; FOP: foramen oval permeable; GEM: gradiente espacial máximo; ICP: intervención coronaria percutánea; LV/RVAD: dispositivo de asistencia del ventrículo izquierdo/ventrículo derecho; PVL: fuga paravalvular; SAR: tasa de absorción específica; TAVI: implante percutáneo de válvula aórtica; TMVR: reparación percutánea de la válvula mitral.
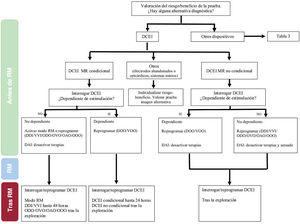
El flujo de trabajo que considerar para los pacientes con dispositivos cardiovasculares que se someten a un estudio de RM dependerá del tipo de dispositivo implantado (figura 1).

Se incluyen en este grupo dispositivos coronarios, vasculares y valvulares, entre otros. Las recomendaciones pueden consultarse en la tabla 3.
Dispositivos cardiovasculares electrónicos implantablesEl flujo de trabajo para la realización de un estudio de RM en un paciente portador de DCEI depende de 3 criterios: a) la condicionalidad del sistema implantado incluyendo todos sus anejos funcionantes o disfuncionantes; b) las condiciones de seguridad del propio paciente definidas fundamentalmente por su carácter de dependiente o no dependiente de la estimulación y el riesgo de arritmias ventriculares, y c) la necesidad del estudio mediante RM en relación con la condición clínica del paciente y la existencia o carencia de técnicas diagnósticas alternativas.
En todos ellos será necesaria la interrogación previa del dispositivo para evaluar la dependencia del paciente de la estimulación y el ajuste de la programación antibradicardia y antitaquicardia, así como la interrogación posterior para confirmar su correcto funcionamiento y hacer la reprogramación correspondiente. En la figura 1 se recoge un algoritmo práctico de actuación en los distintos contextos.
Dispositivos de estimulación, RM condicionalesLas condiciones de compatibilidad con RM de un DCEI vienen definidas por el fabricante, el cual ha sometido el sistema completo a test de validación, teniendo en cuenta que estas pruebas se han realizado con la combinación de las distintas partes del sistema (electrodos y generador) pertenecientes a un mismo fabricante. Por ficha técnica, las condiciones de compatibilidad únicamente se aseguran en estas circunstancias; en el momento actual y a efectos legales, los fabricantes no aseguran la compatibilidad cuando se combinan materiales de distintos fabricantes, si bien no se ha demostrado en la literatura médica que esto suponga un mayor riesgo de complicaciones. Otro factor para tener en cuenta es la existencia de materiales abandonados, disfuncionantes o de implantación epicárdica. En estas condiciones, ningún fabricante asegura el carácter de compatibilidad con RM de sus sistemas.
Por otro lado, la dependencia de la estimulación del paciente se establece ante la ausencia de ritmos intrínsecos o de escape que aseguren un adecuado gasto cardiaco tras el cese de la estimulación. Como regla general, la Sociedad Europea de Cardiología (ESC) establece un límite de 50 lpm para considerar la dependencia del paciente9.
Personal cualificado debe evaluar y revisar a los pacientes y sus respectivos sistemas antes de acudir a la exploración de RM y después de completar el estudio, y debe quedar constancia de ambas actuaciones9. Dado que las condiciones de seguridad se consideran óptimas en estas circunstancias, existe mayor grado de flexibilidad en los modos de programación recomendados y los tiempos recomendados para las interrogaciones.
Antes de la RM, se precisa la evaluación del paciente (comprobando que no tenga electrodos abandonados, epicárdicos, disfuncionantes o conectores extra/adaptadores entre electrodo y generador) e interrogación del dispositivo, así como la programación del modo de compatibilidad de RM si este está disponible entre las posibilidades de programación del dispositivo evaluado. Se precisa una buena comunicación entre el personal de RM y cardiología para asegurar que el sistema es compatible y el paciente está correctamente evaluado y preparado para proceder con la exploración diagnóstica.
Como norma general, para los pacientes con ritmo propio, se recomienda la reprogramación en modo DDI/VVI, lo que permite al paciente portar un dispositivo de estimulación plenamente funcionante durante el estudio9. Además, esto permitiría un mayor margen de tiempo para la reprogramación del dispositivo tras la RM; se recomienda reprogramar en las 48 h posteriores al estudio. Como desventaja, estos modos de programación siguen siendo sensibles a las interferencias por ruidos externos y podrían desencadenar la entrada en funcionamiento de los algoritmos de reversión por ruido, lo que daría lugar a una autoprogramación en modo asíncrono independiente de la frecuencia de base. Como alternativa se puede plantear la reprogramación en modos ODO/OVO/OAO/OOO9, si bien se debe tener en cuenta que en estas condiciones se pierden las funcionalidades de estimulación del dispositivo, por lo que la reprogramación habrá de efectuarse inmediatamente tras el estudio de RM.
En el caso de pacientes dependientes de estimulación con dispositivos RM condicionales y sin función DAI, no se considera necesaria la revisión inmediata tras el procedimiento, pero sí se recomienda hacerla en las primeras 24 h. El modo de estimulación recomendado en estas condiciones es DOO/VOO, con una frecuencia de estimulación 20 lpm superior a la frecuencia intrínseca, si la hay, o adaptada a las necesidades hemodinámicas del paciente en caso de que no haya ritmo intrínseco9.
Otros aspectos relativos a la programación de los dispositivos en estas circunstancias se muestran en la tabla 4, haciendo referencia a las salidas de estimulación, los parámetros de sensado y las funciones adicionales de los dispositivos, entre otros.
Recomendaciones practicas sobre los diferentes aspectos de seguridad en pacientes portadores de dispositivos cardiovasculares electrónicos implantables sometidos a estudios de resonancia magnética
| Condición | Recomendación | Comentario |
|---|---|---|
| Interrogación antes de la RM | En todos los casos | Independientemente de la condición del sistema y para hacer una comprobación previa de su estado |
| Disponible modo de RM | Se recomienda su activación | Ajuste de parámetros acorde a las necesidades particulares de cada paciente |
| Modo de estimulación | DOO/VOO | Pacientes dependientes |
| DDI/VVI | Pacientes con ritmo intrínseco (no dependientes) | |
| ODO/OVO/OAO/OOO | Pacientes no dependientes como alternativa al anterior | |
| Parámetros de estimulación | Estimulación bipolar/amplitud 5 V/anchura de impulso 1 ms | Como norma general en todos los dispositivos |
| Parámetros de sensado | Sensado bipolar | Como norma general en todos los dispositivos |
| Funciones adicionales de estimulación | Desactivadas | Respuesta a la caída de frecuencia, funciones de optimización de la resincronización (respuesta a la fibrilación auricular conducida, disparo por sensado de extrasístoles, etc.), terapia antitaquicardia auricular, etc. |
| Funciones antitaquicardia de DAI | Desactivadas | Desactivar sensado y terapias de taquicardia ventricular/fibrilación ventricular |
| Acompañamiento y monitorización del paciente durante la RM | Monitorización ECG/pulsioximetría | En todos los casos |
| Disponibilidad de equipo para resucitación avanzada | En todos los casos | |
| Personal sanitario en sala con capacidad de asistencia inmediata y soporte vital | En todos los casos | |
| Personal cualificado en sala con capacidad de programación inmediata del dispositivo | En caso de sistemas no condicionales con RM y pacientes dependientes | |
| Personal cualificado en el entorno hospitalario con capacidad de programación inmediata del dispositivo | En caso de sistemas no condicionales con RM y pacientes no dependientesAcorde a la política del centro en el caso de sistemas condicionales con RM | |
| Reprogramación tras la RM | Inmediata | Sistemas no condicionales con RM; sistemas programados a modo ODO/OVO/OAO/OOO; sistemas DAI |
| Dentro de las 24 h | Pacientes dependientes con sistemas de estimulación condicionales con RM y modo de estimulación DOO/VOO | |
| Dentro de las primeras 48 h | Pacientes no dependientes con sistemas de estimulación condicionales con RM y modo de estimulación DDI/VVI | |
| Seguimiento evolutivo | Al cabo de 1 semana. Valorar la posibilidad de efectuarlo mediante seguimiento a distancia | Interrogación general del estado del sistema |
DAI: desfibrilador automático implantable; RM: resonancia magnética.
Para los pacientes portadores de DAI RM condicional, la activación del modo de seguridad RM desactiva las terapias ventriculares antitaquicardia. Esta situación coloca al paciente en un estado de vulnerabilidad frente a las arritmias ventriculares que pudieran ocurrir durante la práctica del estudio de la RM y los tiempos periprocedimiento2.
Dispositivos de estimulación en RM no condicional o electrodos abandonadosEste apartado incluye a los pacientes con electrodos abandonados, sistema (electrodo y/o generador) de RM no condicional, electrodos epicárdicos o conectores extra entre el electrodo y el generador.
Existen estudios que demuestran que en casos necesarios se puede realizar una RM a estos pacientes con seguridad y pocos efectos adversos. Tanto el paciente como el médico que solicita la RM deben de ser informados sobre el tipo de dispositivo y la ausencia de compatibilidad con RM, de los riesgos potenciales para el paciente y la posterior disfunción del dispositivo. Con todo esto se deberá valorar el balance riesgo-beneficio para determinar si es necesario realizar la RM o se puede sustituir por otra prueba de imagen.
Si tras toda la información aportada se considera que la RM es necesaria y debe realizarse, los dispositivos de estimulación RM no condicionales deberán ser evaluados y revisados antes e inmediatamente después del estudio de RM para comprobar su correcto funcionamiento. La programación de estos dispositivos se resume en la tabla 4.
Los dispositivos con función DAI también deberán ser evaluados y revisados antes e inmediatamente después del estudio de RM, en este caso para reprogramar a modo activo las funciones de sensado y tratamiento antitaquicardia2. En los DAI, deben desactivarse todas las terapias antitaquicardia, lo que deja al paciente en un estado de vulnerabilidad frente a las arritmias ventriculares que pudieran ocurrir durante la práctica del estudio de la RM y los tiempos periprocedimiento2.
Por otro lado, las condiciones particulares de este grupo de pacientes y dispositivos los sitúan en una posición de vulnerabilidad, lo que conlleva la necesidad de pautas más estrictas de monitorización y vigilancia durante los estudios de RM. Las recomendaciones generales a este respecto, acordes con las recomendaciones más actuales de la ESC, se muestran en la tabla 4. Respecto al seguimiento evolutivo, la ESC recomienda comprobar la integridad del sistema en el curso de 1 semana. Si bien no existen datos en la literatura que avalen la monitorización a distancia para este fin, es opinión de este comité que se trata de una clara alternativa a la revisión presencial.
Balance riesgo-beneficioSe debe evaluar el balance riesgo-beneficio ante todo paciente con indicación clínica de un estudio de RM que sea portador de un dispositivo cardiovascular. Como se ha comentado, debe considerarse el tipo de dispositivo, su compatibilidad con la RM, la dependencia del ritmo de estimulación del paciente y la necesidad clínica de realizar el estudio de RM, así como si se puede llegar al diagnóstico mediante una técnica de imagen alternativa.
Con las debidas precauciones, la presencia de estos dispositivos no debería ser una limitación para una RM con indicación clínica establecida o urgente. En este sentido, el estudio MagnaSafe demostró que serían seguros los estudios de RM no torácica en equipos de 1,5 T para pacientes portadores de DAI o marcapasos no condicionales para RM que hayan sido programados adecuadamente antes de la exploración24.
En términos generales, hay más evidencia sobre la compatibilidad de sistemas en equipos de 1,5 T que en equipos de mayor campo magnético. Por lo tanto, y si de ello no deriva un estudio de calidad no diagnóstica, sería preferible la utilización de equipos de 1,5 T.
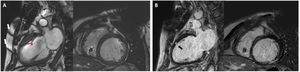
MEJORA Y OPTIMIZACIÓN DE LA IMAGEN DE RM CARDIOTORÁCICA DE PACIENTES PORTADORES DE DISPOSITIVOSEn un estudio de RM, el tipo de dispositivo y su localización constituyen los principales condicionantes de la calidad de la imagen. En RM cardiotorácica, los DAI y la terapia de resincronización cardiaca suelen producir más artefactos por su mayor volumen y la utilización de elementos ferromagnéticos, y puede observarse distorsión de la imagen hasta a 12cm del generador (figura 2). Por esta razón, los dispositivos implantados en el lado izquierdo crean más artefactos que los localizados en el lado derecho47, que en general no afectan a la imagen cardiaca48.
 se observan artefactos en la pared torácica (flechas blancas) que afectan a la región anterior (asterisco) en la imagen de 2 cámaras; sin embargo permite la medición en la imagen en eje corto. B: en las imágenes de realce tardío (GSE-IR, gradient spoiled echo inversion-recovery), el artefacto es menos marcado y se puede valorar correctamente en ambas geometrías un área de realce tardío (flecha negra).")
Imágenes obtenidas con equipo de RM de 1,5 T en un paciente portador de marcapasos. A: en secuencia cine SSFP (steady-state free precession) se observan artefactos en la pared torácica (flechas blancas) que afectan a la región anterior (asterisco) en la imagen de 2 cámaras; sin embargo permite la medición en la imagen en eje corto. B: en las imágenes de realce tardío (GSE-IR, gradient spoiled echo inversion-recovery), el artefacto es menos marcado y se puede valorar correctamente en ambas geometrías un área de realce tardío (flecha negra).
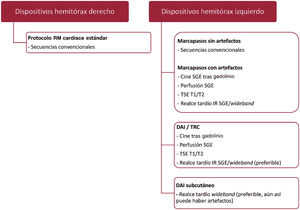
El protocolo de exploración de RM cardiaca debe dirigirse a contestar la pregunta del clínico, limitarlo a las secuencias imprescindibles y no debe alargar el estudio innecesariamente (figura 3).
/banda ancha; RM: resonancia magnética; SGE: spoiled echo-gradient (eco de gradiente distorsionado); TRC: terapia de resincronización cardiaca; TSE: turbo spin-echo (eco de spin turbo).")
Algoritmo práctico de optimización de la imagen de pacientes portadores de dispositivos cardiovasculares electrónicos implantables sometidos a estudios de RM cardiotorácica. DAI: desfibrilador automático implantable; realce tardío IR SGE/wideband: realce tardío inversión-recuperación spoiled echo-gradient (eco de gradiente distorsionado)/banda ancha; RM: resonancia magnética; SGE: spoiled echo-gradient (eco de gradiente distorsionado); TRC: terapia de resincronización cardiaca; TSE: turbo spin-echo (eco de spin turbo).
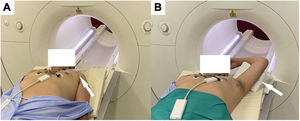
En general, para reducir los artefactos secundarios a DCEI, se debe intentar un aumento de la distancia entre el dispositivo y el área de exploración. Algunas maniobras útiles son colocar por encima de la cabeza el brazo homolateral al generador o adquirir la imagen en inspiración profunda (figura 4).
 o con el brazo homolateral al dispositivo colocado hacia arriba (B).")
Preparación de paciente portador de dispositivo automático implantable en hemitórax izquierdo para una RM cardiotorácica. Aumento de la distancia entre el generador y el área de exploración mediante la colocación de una venda adhesiva (A) o con el brazo homolateral al dispositivo colocado hacia arriba (B).
En relación con la selección de protocolos y secuencias de exploración, cabe destacar el uso reciente de las secuencias de realce tardío de banda ancha (wideband) para reducir los artefactos asociados con DCEI, si bien aún no están clínicamente disponibles en muchos centros. La tabla 5, la figura 3 y la figura 4 recogen esta y otras recomendaciones específicas en relación con las secuencias de exploración en un estudio de RM cardiaca.
Recomendaciones para mejorar la imagen cardiotorácica según las secuencias de resonancia magnética utilizadas
| Tipo de secuencia | Recomendaciones y ajustes específicos | Imágenes de ejemplo |
|---|---|---|
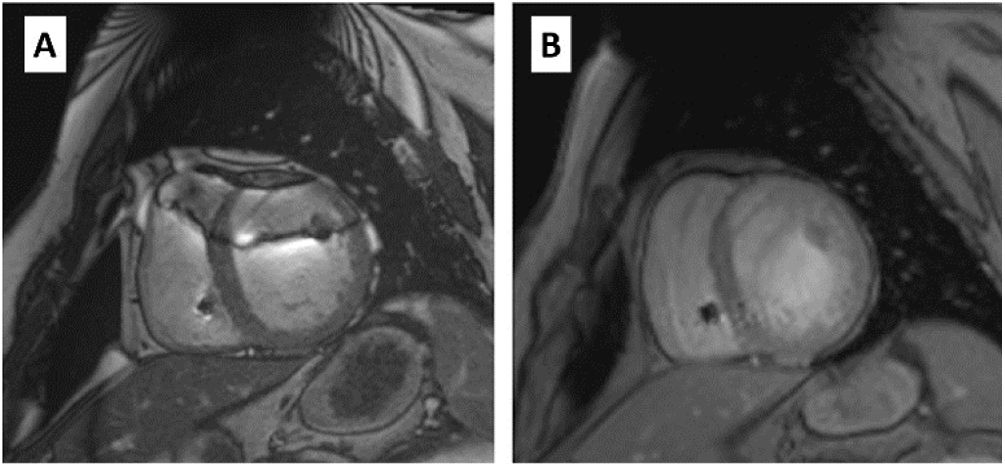
| Cine | Si hay artefacto importante en imágenes SSFP, la secuencia SGE puede mejorar la imagen. Intentar utilizar CS y reducción del TE49,50La adquisición de cine poscontraste permite una mejor detección del endocardio ventricular51Si se usan secuencias de cine SSFP, valorar: a) utilizar explorador de frecuencia para elegir dónde los artefactos son menos importantes; b) aumentar el ancho de banda y disminuir ligeramente la resolución para reducir el TR al mínimo posible50 | DAI implantado en lado izquierdo. A: cine-SSFP con extenso artefacto de bandas de resonancia. B: cine-SGE en la misma posición de exploración |
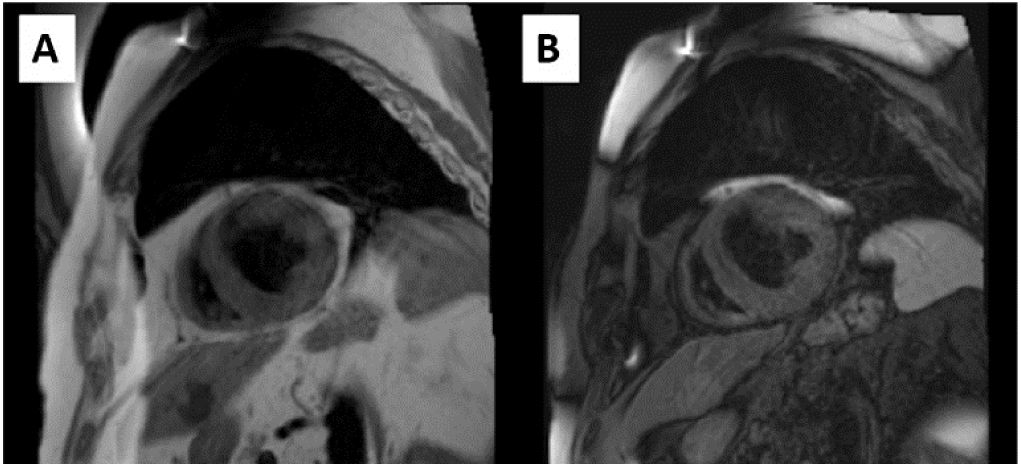
| Caracterización tisular T1 y T2 | En general, las imágenes de sangre negra TSE potenciadas en T1 y T2 son menos sensibles a los artefactos producidos por estos dispositivos, y se obtiene buena calidad de imagen48 | DAI izquierdo. A: TSE-T1w eje corto sin artefacto. B: TSE-T2w eje corto sin artefacto. Equipo 1,5 T |
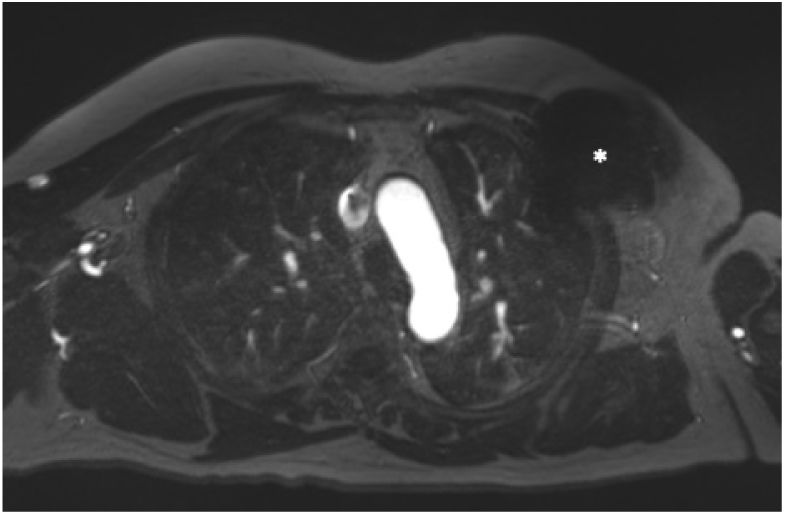
| Perfusión y angiografía 3D | Las secuencias de perfusión SGE presentan menos artefactos que las SSFP51Angiografía 3D: secuencias libres de artefactos. Permiten una buena visualización de los grandes vasos y sus ramas secundarias48,51 | Angiografía 3D: imagen axial potenciada en T1 con escasos artefactos salvo en la región más cercana al generador |
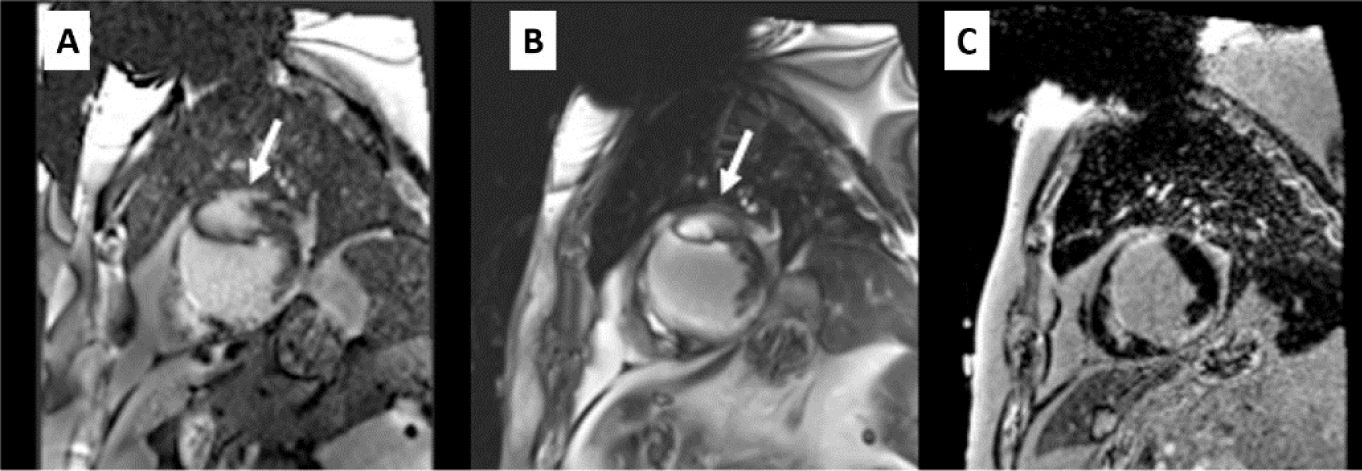
| Realce tardío | Las secuencias de adquisición rápida (single-shot) SSFP (útiles en ritmo irregular o imposibilidad de apnea prolongada) son más propensas a los artefactos, por lo que no estarían recomendadasLas SGE de IR pueden ocasionar artefactos de hiperintensidad o vacío de señal. Si están disponibles, se recomienda ampliar el ancho de banda mediante la utilización de IR-WB, que permite reducir o desplazar el artefacto fuera del campo de exploración52Antes de IR-WB, es recomendable una exploración previa a la administración de contraste en geometrías de 4 y 2 cámaras con 3 frecuencias diferentes (–1.500, 0 y +1.500 Hz), con el fin de determinar la frecuencia en la que es menos probable que aparezca el artefacto de hiperintensidad. Dicha frecuencia óptima se aplicará en la adquisición posterior de IR-WB53El cambio de frecuencia óptimo depende de factores del paciente, como la lateralidad y el tipo de dispositivo. En los pacientes con implante en el lado derecho, el artefacto es mínimo tanto en las imágenes estándar como en WB. En pacientes con implantes izquierdos, se puede minimizar el artefacto con secuencia WV en marcapasos y DAI. Sin embargo, en terapia de resincronización cardiaca y dispositivos subcutáneos tiene un impacto menor, observándose un artefacto de vacío de señal en la pared lateral con mayor frecuencia53 | DAI izquierdo. A: single-shot SSFP (artefacto, flecha). B: SGE-IR (artefacto, flecha). C: IR-WB 6 kHz sin artefactosDAI subcutáneo izquierdo. A: localizador coronal, artefacto pared lateral (estrella). B: IR-WB tras múltiples ajustes persiste artefacto pared lateral. C: IR-WB dos cámaras, único plano sin artefacto |

3D: tridimensional; CS: compressed sensing (detección comprimida); DAI: desfibrilador automático implantable; IR: inversión-recuperación; SGE: spoiled gradient-echo (eco de gradiente distorsionado); SSFP: steady-state free precession (estado de equilibrio libre de precesión); TE: tiempo de eco; TR: tiempo de repetición; TSE: turbo spin-echo (eco de spin turbo); WB: wideband (banda ancha).
La realización de estudios de RM a pacientes portadores de dispositivos cardiovasculares es posible con las debidas precauciones. El presente documento ofrece una revisión y pautas de manejo para evitar la interferencia del campo magnético en el dispositivo electrónico y reducir los artefactos de imagen generados con el dispositivo cardiovascular en el área de exploración cardiotorácica.
FINANCIACIÓNNinguna.
CONTRIBUCIÓN DE LOS AUTORESTodos los autores han participado por igual en la elaboración de este documento. El presente documento ha sido revisado antes de su publicación por expertos externos a su redacción: J.F. Rodríguez Palomares, J.M. Tolosana, J.A. Hidalgo Pérez, E. Pérez-David, Vicente Bertomeu-González y H. Cuéllar.
CONFLICTO DE INTERESESNinguno.
Grupo de Trabajo de Cardiorresonancia Magnética y Cardiotomografía Computarizadas de la Sociedad Española de Cardiología (SEC-CRMTC): M. Barreiro-Pérez, A. Maceira González, S. Prat-González, L.J. Jiménez-Borreguero y C. Fernández-Golfín Lobán.
Asociación del Ritmo Cardiaco de la Sociedad Española de Cardiología (SEC- Asociación del Ritmo Cardiaco): D. Calvo y T. Datino.
Sociedad Española de Radiología Médica (SERAM) y Sociedad Española de Imagen Cardiotorácica (SEICAT): B. Cabeza, J.L. Reyes-Juárez, E. Vañó Galván, C. Delgado Sánchez-Gracián, R.J. Perea, G. Bastarrika y M. Sánchez.
Grupo de Trabajo de Cardio RM y Cardio TC de la Sociedad Española de Cardiología (SEC-CRMTC): J.F. Rodríguez-Palomares y E. Pérez-David.
Asociación del Ritmo Cardiaco de la Sociedad Española de Cardiología (SEC- Asociación del Ritmo Cardiaco): J.M. Tolosana y V. Bertomeu-González.
Sociedad Española de Radiología Médica (SERAM) y Sociedad Española de Imagen Cardiotorácica (SEICAT): J.A. Hidalgo Pérez y H. Cuéllar.