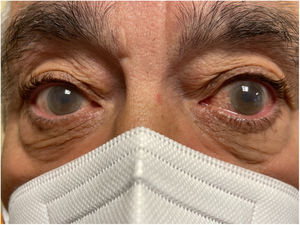

La hipoalfalipoproteinemia primaria es un trastorno infrecuente caracterizado por la producción alterada de apolipoproteína (apo) A-I, cuyo resultado son cifras marcadamente bajas de colesterol unido a lipoproteínas de alta densidad. La imagen corresponde a un paciente de 60 años, hijo de padres consanguíneos, con concentraciones de lipoproteínas de alta densidad de 7mg/dl (40-60), triglicéridos en 135mg/dl, colesterol total en 119mg/dl, lipoproteínas de baja densidad en 83mg/dl, lipoproteína a en 15,8mg/dl (< 30) y apoB en 99mg/dl (55-140). Estaba en tratamiento con simvastatina 10mg diarios.
En la exploración destacaron xantelasmas y arcos corneales completos con opacidades corneales bilaterales que no condicionan la agudeza visual (figura 1). El paciente dio por escrito su consentimiento para la realización de las fotografías y la publicación del caso. La sospecha diagnóstica inicial fue una deficiencia de lecitina-colesterol-acil-transferasa, ya que las opacidades corneales se relacionan clásicamente con esta afección, cuya forma parcial se conoce como «enfermedad del ojo de pescado». Se solicitó un examen genético, que halló una mutación en homocigosis en el gen de la apoA-1 asociado con hipoalfalipoproteinemia primaria (NM_000039.3(APOA1):c.67C>T (p.Gln23*)). Para un correcto diagnóstico es imprescindible un examen genético. Actualmente, el abordaje consiste en el control de las cifras de lipoproteínas de baja densidad y el pronóstico va a estar determinado por la aparición prematura de enfermedad ateroesclerótica.
FINANCIACIÓN
No se ha recibido ningún tipo de financiación para la redacción y la publicación de este trabajo.
CONTRIBUCIÓN DE LOS AUTORESJ. Costas Eimil: redacción y diseño. P. Sánchez-Sobrino: revisión y aprobación.
CONFLICTO DE INTERESESLos autores declaran no tener ningún conflicto de intereses.