

Recientemente se han descrito mutaciones missense en la filamina C (FLNC) como causa de miocardiopatía. Los conocimientos sobre la patogenicidad y la correlación genotipo-fenotipo son escasos. Nuestro objetivo es describir un fenotipo cardiaco distintivo relacionado con mutaciones missense en el dominio ROD2 de FLNC (FLNC-mRod2).
MétodosIncluimos 21 familias independientes con fenotipo de miocardiopatía hipertrófica (MCH)/miocardiopatía restrictiva (MCR) portadoras de variantes missense en FLNC-mRod2. Se estudió clínicamente a los portadores, además de hacer un cribado en cascada. Se analizó histológicamente el tejido miocárdico de tres corazones explantados y se comparó con un corazón portador de un truncamiento de FLNC y con un control sano. Se transfectaron plásmidos con mutaciones missense de FLNC y se analizaron mediante microscopía confocal.
ResultadosEn 11 familias (52%) con 20 individuos evaluados (37 [23,7-52,7] años), 15 casos presentaron un fenotipo cardiaco consistente en una superposición de MCH-MCR e hipertrabeculación ventricular izquierda (apariencia de dientes de sierra). Durante una mediana de seguimiento de 6,49 años presentaron principalmente insuficiencia cardiaca avanzada (16 (80%) disfunción diastólica, 3 trasplantes cardiacos, 3 muertes por insuficiencia cardiaca) en ausencia de alteraciones de la conducción cardiaca o miopatía esquelética. Un total de 6 familias presentaban segregación genotipo-fenotipo leve, y las restantes eran mutaciones de novo. Se observó una remodelación de la matriz extracelular y distribución de la FLNC diferencial en los cardiomiocitos. Las células HT1080 y H9c2 no revelaron agregados citoplasmáticos de FLNC.
ConclusionesLas variantes en FLNC-mRod2 exhiben una alta prevalencia de fenotipo solapado de MCR, MCH e hipertrabeculación en dientes de sierra, con una remodelación histopatológica cardiaca distintiva.
Palabras clave
La creciente disponibilidad de la tecnología de secuenciación de nueva generación ha conducido al importante conocimiento de que las miocardiopatías son trastornos heterogéneos con expresividad y penetrancia variables. En la mayor parte de las miocardiopatías, los mecanismos de la relación entre genotipo y fenotipo dentro del mismo gen mutado continúan siendo en gran parte desconocidos.
El gen FLNC codifica la importante proteína cardiaca filamina C, que se expresa en las células del músculo esquelético e interactúa con varias proteínas en el disco Z de los sarcómeros y con las funciones estructurales y de señalización en el miocardiocito. El gen contiene una región de unión a la actina, 2 regiones bisagra y un dominio con 24 repeticiones de tipo Ig. La FLNC participa en la miogénesis preservando las miofibrillas y la ultraestructura sarcomérica. Además, la FLNC actúa como proteína de membrana de anclaje para las proteínas citoesqueléticas estructurales1,2.
Clásicamente, las mutaciones del gen FLNC se han asociado a un amplio espectro de enfermedades musculares hereditarias a las que se denomina miopatías miofibrilares3. En la última década, unos pocos estudios con un pequeño número de pacientes han descrito variantes missense del gen FLNC que causan miocardiopatía restrictiva (MCR) hereditaria4-6, miocardiopatía hipertrófica (MCH)7 o miocardiopatía no compactada (MCNC)8 con una ausencia casi completa de afección del músculo esquelético. Más recientemente, varios estudios han descrito variantes truncadas en el gen FLNC (FLNC-tv) relacionadas con una miocardiopatía arritmógena9,10. A pesar de su creciente identificación como causa genética de miocardiopatías hereditarias, la relación causal entre las variantes missense de FLNC y los fenotipos cardiacos solapados no se ha establecido claramente.
Nuestro objetivo es describir las características fenotípicas de la miocardiopatía relacionada con variantes missense del gen FLNC en el dominio ROD2 (FLNC-mRod2).
MÉTODOSPoblación del estudio y evaluación clínicaSe incluyó a todos los probandos que habían sido remitidos para una evaluación genética cardiovascular en el mismo laboratorio de referencia a causa de una MCH o una MCR, presentaban una variante de FLNC-mRod2 y estaban disponibles para el estudio. Se excluyó a todos los individuos con otra variante causal que explicara el fenotipo. Después de la primera evaluación, se seleccionó a las familias con miocardio en dientes de sierra en la exploración de imagen cardiovascular en un estudio clínico detallado.
Se llevó a cabo una evaluación clínica exhaustiva, que incluyó una evaluación detallada de la historia clínica del paciente, el árbol genealógico, la creatincinasa, un electrocardiograma de 12 derivaciones, una monitorización Holter y una evaluación ecocardiográfica por un experto. La disfunción de la fracción de eyección del ventrículo izquierdo (FEVI) se evaluó con el método de Simpson biplano11. La función diastólica se evaluó según las recomendaciones actuales12. Se analizó el aspecto del miocardio del ventrículo izquierdo (VI) prestando especial atención a la hipertrabeculación, según los criterios diagnósticos actuales para la MCNC con 2 capas bien definidas (capa delgada compactada y capa gruesa no compactada)13. El miocardio en dientes de sierra se identificó por la presencia de múltiples proyecciones e invaginaciones profundas en un miocardio denso y compactado.
El estudio fue aprobado por el comité de ética de investigación local. Se obtuvo el consentimiento informado por escrito para la inclusión en el estudio de todos los participantes o, en el caso de pacientes menores de edad o personas fallecidas, de los familiares de primer grado.
Secuenciación genéticaEn el mismo centro se obtuvieron muestras de sangre de los probandos para la secuenciación genética mediante un panel de secuenciación de nueva generación orientado por el fenotipo. En el caso de los probandos con solapamiento o un fenotipo no definido, se empleó un panel de secuenciación de nueva generación que incluía genes relacionados con la cardiopatía o genes candidatos. Se capturaron los exones codificadores y los límites intrónicos de 247 genes relacionados con enfermedades cardiovasculares hereditarias (tabla 1 del material adicional) utilizando una biblioteca de sondas específica.
Para la evaluación de la patogenicidad de las variantes genéticas, se consideraron datos como la frecuencia en las bases de datos públicas (por ejemplo, Human Gene Mutation Database, Single Nucleotide Polymorphism Database, NHLBI GO Exome Sequencing Project y ClinVar o Genome Aggregation Database14–16), su descripción previa en la literatura y la conservación entre las especies. Después de la identificación de las variantes raras del gen FLNC en los pacientes índice, se llevó a cabo sistemáticamente un examen de detección genética en cascada en todos los familiares disponibles mediante secuenciación de ADN Sanger. Para las variantes de novo, se confirmó la paternidad cuando fue posible. Por último, se determinó el logaritmo de la puntuación de probabilidad17 y se determinó la clasificación del American College of Medical Genetics and Genomics (ACMG)18. Puede consultarse información detallada sobre el método para el análisis genético en el material adicional.
Evaluación histológicaSe llevó a cabo una caracterización histológica, histoquímica e inmunohistoquímica completa del tejido cardiaco procedente de 3 corazones explantados (paciente de la familia F y familia C, pacientes 2 y 3). Las muestras de tejido se fijaron durante al menos 48 h en formaldehído tamponado neutro al 10%, se lavaron, se deshidrataron con concentraciones crecientes de etanol, se aclararon en xilol y finalmente se incluyeron en parafina según un protocolo convencional19.
Se obtuvieron cortes histológicos de un grosor de 5 μm, que se desparafinaron, se hidrataron y se examinaron en los siguientes pasos: a) se realizó un examen histológico general con hematoxilina-eosina; b) se tiñeron histoquímicamente las miofibrillas de los miocardiocitos con hematoxilina de Heidenhain; c) se evaluó la organización y distribución de los componentes principales de la matriz extracelular (MEC) con la tinción de picrosirio del colágeno, la técnica de reducción de metal de Gomori para las fibras reticulares, la tinción histoquímica de orceína para las fibras elásticas y la tinción de azul alcián para los proteoglicanos. Además, se identificaron los colágenos de tipo I y IV (COL-I y COL-IV respectivamente) mediante inmunohistoquímica, y d) se determinó el patrón de distribución del FLNC y la conexina-43 (Cx-43) mediante inmunohistoquímica. La información técnica sobre los anticuerpos utilizados se resume en la tabla 2 del material adicional.
Además de la caracterización histológica, se llevó a cabo un análisis semicuantitativo del cociente célula/colágeno del área de MEC en 3 cortes diferentes procedentes de 3 preparaciones tisulares distintas, con el método de picrosirio del programa informático Image J (National Institute of Health, Estados Unidos) y siguiendo un procedimiento descrito anteriormente20. Además, se seleccionaron 20 puntos en 3 cortes de cada condición teñidos con el método de hematoxilina de Heidenhain y se determinó la intensidad de la reacción histoquímica para las miofibrillas. Por último, se calculó la anchura celular en 20 células por imagen, seleccionadas en 3 imágenes de cada trastorno.
Para detectar las alteraciones histológicas específicas atribuibles a las variantes de FLNC-mRod2, se compararon las observaciones histológicas con una muestra de tejido cardiaco no patológico (control) y con una muestra de un portador de una FLNC-tv procedente de nuestra cohorte histórica. El caso de FLNC-tv mostró el fenotipo solapado reconocido de miocardiopatía dilatada y miocardiopatía arritmógena9,10.
Generación de plásmidos, cultivo celular y microscopia confocalSe utilizó el plásmido de tipo natural o salvaje (wild-type) pCMV6-FLNC (212462; OriGene Technologies, Estados Unidos) como plantilla para insertar las variantes p.P2301L, p.E3224K y p.R2340W, utilizando el kit Quik Change Lightning (Agilent Technologies, Estados Unidos) en combinación con los oligonucleótidos apropiados (tabla 3 del material adicional). Se verificó el FLNC que codificaba el ácido desoxirribonucleico complementario (ADNc) de todos los plásmidos con el método de secuenciación Sanger (Macrogen, Países Bajos). Se cultivaron células HT1080 (DSMZ, German Collection of Microorganisms and Cell Culture, ACC315, Alemania) y H9C2 (ATCC, CRL-1446, Estados Unidos) en cámaras μSlide (ibidi GmbH, Alemania) utilizando el medio de cultivo Modified Eagle Medium de Dulbecco suplementado con suero de ternera fetal al 10% y penicilina/estreptomicina. Se utilizó Lipofectamin 3000 (Thermo Fisher Scientific, Estados Unidos) para la transfección celular según las instrucciones del fabricante. A las 24 h de la transfección, se lavaron las células con solución salina tamponada con fosfato y se fijaron durante 10min a temperatura ambiente utilizando Histofix al 4% (Carl-Roth, Alemania). El análisis inmunocitoquímico se llevó a cabo, según lo recientemente descrito, mediante el microscopio confocal TCS SP8 (Leica Microsystems, Alemania)4.
Análisis estadísticoPara los análisis estadísticos se empleó el programa SPSS Statistics, versión 21.0 (IBM Corp, Estados Unidos). Las variables continuas se presentan en forma de media ± desviación estándar para cada parámetro. Para la cuantificación de las variables histológicas, se aplicó la prueba de la U de Mann-Whitney (no paramétrica). Todas las variables cualitativas se expresan mediante frecuencia y porcentaje.
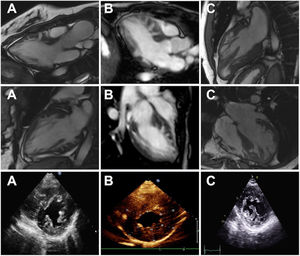
RESULTADOSCaracterísticas de los pacientesSe incluyó a 21 familias no relacionadas, procedentes de 7 centros de referencia, a las que se examinó genéticamente respecto a la MCH/MCR y en las que se encontró que eran portadoras de variantes raras de FLNC-mRod2 (figura 1 del material adicional). Un total de 11 familias (52%) con 20 individuos (37 [23,7-52,7] años) mostraron 15 casos con un fenotipo cardiaco consistente en una MCH o una MCR, con una hipertrabeculación del ventrículo izquierdo (HTVI) marcada y atípica que distorsionaba el VI, con presencia de numerosos puentes cruzados de proyecciones musculares. Estas protrusiones de puentes musculares en el VI delineaban criptas profundas en un único miocardio denso y compactado (figura 1), que se ha identificado como aspecto en «dientes de sierra», diferente de las 2 capas características de la MCNC (capa fina compactada y capa gruesa no compactada). Además, esta HTVI singular se confirmó en 3 corazones explantados (figura 2).
 y de ecocardiografía (recuadro inferior) que muestran el aspecto de la hipertrabeculación del ventrículo izquierdo en dientes de sierra con proyecciones musculares y criptas profundas que distorsionan la arquitectura normal del ventrículo izquierdo. A: paciente III.1 de la familia C. B: paciente II.2 de la familia A. C: paciente de la familia E.")
Imágenes de resonancia magnética cardiaca (recuadros superior y central) y de ecocardiografía (recuadro inferior) que muestran el aspecto de la hipertrabeculación del ventrículo izquierdo en dientes de sierra con proyecciones musculares y criptas profundas que distorsionan la arquitectura normal del ventrículo izquierdo. A: paciente III.1 de la familia C. B: paciente II.2 de la familia A. C: paciente de la familia E.
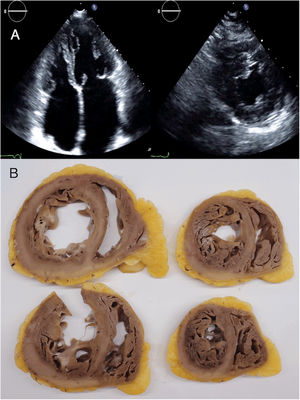
 e imágenes macroscópicas del corazón explantado (B) del paciente índice de la familia C, de 24 y 36 años respectivamente. Las imágenes de ecocardiografía mostraron protrusiones musculares en el ventrículo izquierdo. Las imágenes anatomopatológicas mostraron la hipertrabeculación atípica intensa, con recesos profundos.")
Imágenes de ecocardiografía (A) e imágenes macroscópicas del corazón explantado (B) del paciente índice de la familia C, de 24 y 36 años respectivamente. Las imágenes de ecocardiografía mostraron protrusiones musculares en el ventrículo izquierdo. Las imágenes anatomopatológicas mostraron la hipertrabeculación atípica intensa, con recesos profundos.
En la cohorte total de 20 individuos, la mediana de edad en el momento del diagnóstico era 37 [23,7-52,7] años y 12 (60%) eran varones. La MCH fue más frecuente (13; 65%) que la MCR (4; 20%), y 15 individuos (75%) mostraron un aspecto en dientes de sierra. Un total de 16 individuos (80%) presentan una disfunción diastólica marcada (grado II o III) y en 10 (50%) había una fibrilación auricular, a veces en la adolescencia o al inicio de la edad adulta.
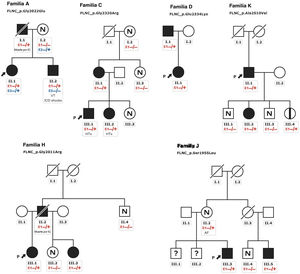
Los pacientes con MCH presentaban un aumento del tabique interventricular (14,3±7mm), una aurícula izquierda dilatada (45,6±10,4mm) y una FEVI inicialmente conservada (58,5±10,3%). Se halló una válvula aórtica bicúspide en 2 individuos de la familia A y 2 de la familia C. Además, se realizó una resonancia magnética cardiaca con evaluación del realce tardío de gadolinio a 9 pacientes, pero ninguno mostró una cicatriz con realce tardío de gadolinio. Todos ellos mostraron un electrocardiograma anormal: en 5 había una inversión de la onda T en derivaciones precordiales y en 8, en derivaciones inferiores y precordiales; 5 tenían un bloqueo de rama derecha del haz de His y en 2 se observó un retraso de la conducción intraventricular inespecífico. Ninguno de los pacientes tenía voltajes bajos ni bloqueo auriculoventricular. La monitorización Holter no identificó un aumento de las extrasístoles ventriculares, pero en 1 individuo identificó una taquicardia ventricular no sostenida. Ninguno de los individuos tenía síntomas, signos o antecedentes de miopatía del músculo esquelético (concentración media de creatincinasa, 137 ± 58 U/l). El cribado genético en cascada para la detección sistemática mostró una cosegregación leve de 6 variantes con el fenotipo (familias A, C, D, H, J, K) (figura 3), mientras que los 5 casos restantes fueron de novo (tabla 1; figura 2 del material adicional y tabla 4 del material adicional). Los no portadores no mostraban manifestaciones de miocardiopatía.
 indica no disponible para la evaluación. Las flechas + P indican el probando. Portadores heterocigotos (E1 −/+) y no portadores (E1 −/−). DAI: desfibrilador automático implantable; IC: insuficiencia cardiaca; TxC: trasplante cardiaco; TVS: taquicardia ventricular sostenida.")
Árboles genealógicos con una cosegregación leve. Los cuadrados indican sexo masculino y los círculos, sexo femenino; las barras inclinadas indican individuos fallecidos, el sombreado negro indica un fenotipo de miocardiopatía, la banda negra indica un portador asintomático, N indica ausencia del fenotipo y (?) indica no disponible para la evaluación. Las flechas + P indican el probando. Portadores heterocigotos (E1 −/+) y no portadores (E1 −/−). DAI: desfibrilador automático implantable; IC: insuficiencia cardiaca; TxC: trasplante cardiaco; TVS: taquicardia ventricular sostenida.
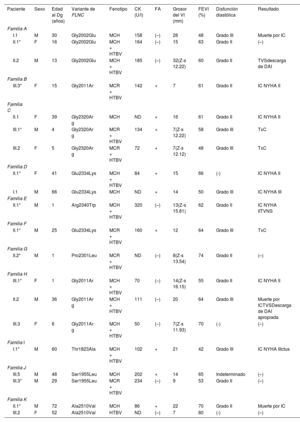
Características clínicas de los portadores evaluados
| Paciente | Sexo | Edad al Dg (años) | Variante de FLNC | Fenotipo | CK (U/l) | FA | Grosor del VI (mm) | FEVI (%) | Disfunción diastólica | Resultado |
|---|---|---|---|---|---|---|---|---|---|---|
| Familia A | ||||||||||
| I.1 | M | 30 | Gly2002Glu | MCH | 158 | (–) | 26 | 48 | Grado III | Muerte por IC |
| II.1* | F | 16 | Gly2002Glu | MCH + HTBV | 164 | (–) | 15 | 63 | Grado II | (–) |
| II.2 | M | 13 | Gly2002Glu | MCH + HTBV | 185 | (–) | 32(Z-s 12.22) | 60 | Grado II | TVSdescarga de DAI |
| Familia B | ||||||||||
| III.3* | F | 15 | Gly2011Ar | MCR + HTBV | 142 | + | 7 | 61 | Grado II | IC NYHA II |
| Familia C | ||||||||||
| II.1 | F | 39 | Gly2320Ar g | MCH | ND | + | 16 | 61 | Grado II | IC NYHA II |
| III.1* | M | 4 | Gly2320Ar g | MCR + HTBV | 134 | + | 7(Z-s 12.22) | 58 | Grado III | TxC |
| III.2 | F | 5 | Gly2320Ar g | MCR + HTBV | 72 | + | 7(Z-s 12.12) | 48 | Grado III | TxC |
| Familia D | ||||||||||
| II.1* | F | 41 | Glu2334Lys | MCH + HTBV | 84 | + | 15 | 66 | (-) | IC NYHA II |
| I.1 | M | 66 | Glu2334Lys | MCH | ND | + | 14 | 50 | Grado III | IC NYHA III |
| Familia E | ||||||||||
| II.1* | M | 1 | Arg2340Trp | MCH + HTBV | 320 | (–) | 13(Z-s 15.81) | 62 | Grado II | IC NYHA IITVNS |
| Familia F | ||||||||||
| II.1* | M | 25 | Glu2334Lys | MCR + HTBV | 160 | + | 12 | 64 | Grado III | TxC |
| Familia G | ||||||||||
| II.2* | M | 1 | Pro2301Leu | MCR + HTBV | ND | (–) | 8(Z-s 13.54) | 74 | Grado II | (–) |
| Familia H | ||||||||||
| III.1* | F | 1 | Gly2011Ar | MCH + HTBV | 70 | (–) | 14(Z-s 16.15) | 55 | Grado II | IC NYHA II |
| II.2 | M | 36 | Gly2011Ar g | MCH + HTBV | 111 | (–) | 20 | 64 | Grado III | Muerte por ICTVSDescarga de DAI apropiada |
| III.3 | F | 6 | Gly2011Ar g | MCH + HTBV | 50 | (–) | 7(Z-s 11.93) | 70 | (-) | (–) |
| Familia I | ||||||||||
| I.1* | M | 60 | Thr1823Ala | MCH + HTBV | 102 | + | 21 | 42 | Grado III | IC NYHA IIIctus |
| Familia J | ||||||||||
| III.5 | M | 48 | Ser1955Leu | MCH | 202 | + | 14 | 65 | Indeterminado | (–) |
| III.3* | M | 29 | Ser1955Leu | MCR + HTBV | 234 | (–) | 9 | 53 | Grado II | (–) |
| Familia K | ||||||||||
| II.1* | M | 72 | Ala2510Val | MCH | 86 | + | 22 | 70 | Grado II | Muerte por IC |
| III.2 | F | 52 | Ala2510Val | HTBV | ND | (–) | 7 | 80 | (-) | (–) |
(–): no presente; (+): presente; CK: creatincinasa (valores de referencia, 11-145 U/l); DAI: desfibrilador automático implantable; Dg: diagnóstico; F: sexo femenino; FA: fibrilación auricular; FEVI: fracción de eyección del ventrículo izquierdo; HTVI: hipertrabeculación del ventrículo izquierdo; IC: insuficiencia cardiaca; M: sexo masculino; MCH: miocardiopatía hipertrófica; MCR: miocardiopatía restrictiva; ND: no disponible; NYHA: clase funcional de la New York Heart Association; TIV: tabique interventricular; TVNS: taquicardia ventricular no sostenida; TVS: taquicardia ventricular sostenida; TxC: trasplante cardiaco; VI: ventrículo izquierdo.
En las 10 familias restantes en las que se identificaron variantes de FLNC-mRod2 sin aspecto en dientes de sierra, había 4 pacientes con MCH obstructiva (1 con HTVI pero sin el rasgo en dientes de sierra), 1 con MCH en el análisis post mortem, 1 con MCH apical, 1 con MCR y 3 individuos sin miocardiopatía.
Eventos clínicosDespués de una mediana de seguimiento de 6,49 [3,8-21,33] años tras el diagnóstico del fenotipo, 11 pacientes (55%) habían sufrido insuficiencia cardiaca (IC) sintomática de clase ≥ II de la New York Heart Association (NYHA), y 3 de ellos necesitaron un trasplante cardiaco debido a la IC avanzada a una edad de entre 30 y 40 años. Tres pacientes (15%) fallecieron por IC. Además, 4 (26,6%) sufrieron un síncope durante el seguimiento y 2, una taquicardia ventricular sostenida (TVS) que motivó el implante de un desfibrilador automático implantable (DAI).
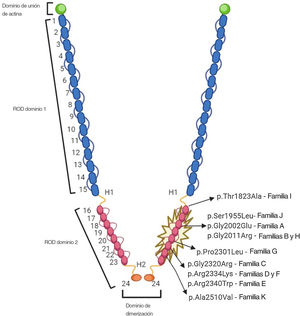
Variantes genéticasEn la figura 4 se muestra la ubicación de las 9 variantes raras identificadas en el dominio de ROD2 de FLNC. Todas las variantes missense observadas en esta cohorte se encontraban en el dominio ROD2, y específicamente en los dominios de tipo Ig 18 a 21. Ninguna de las variantes identificadas estaba presente en las bases de datos de poblaciones de control y los predictores bioinformáticos indicaron que eran probablemente nocivas. La clasificación del ACMG identificó 4 variantes como probablemente patógenas y 5 variantes como de significado desconocido (tablas 5 y 6 del material adicional). No se identificó una segunda variante genética causal relevante en ninguno de los probandos, y se confirmó la paternidad en 4 de los casos de novo. No se detectó ninguna variación en el número de copias (CNV) en la cohorte seleccionada. En todos estos casos, la calidad de la muestra fue suficiente para llevar a cabo este análisis.
Histología, histoquímica e inmunohistoquímica
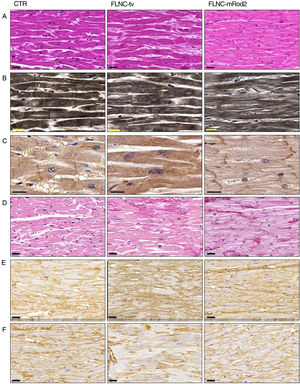
La tinción de hematoxilina-eosina reveló la presencia de células cardiacas moderadamente más grandes en las muestras de corazones con FLNC-tv en comparación con las de corazones con FLNC-mRod2 y las de los controles (figura 5A). También puso de manifiesto una menor densidad de miofibrillas y una ligera reducción del espacio intercelular en los tejidos afectados por FLNC-mRod2, lo cual daba lugar a un patrón de organización del tejido más compacto en estas muestras específicas (figura 5B). Estos resultados se confirmaron mediante análisis histológicos cuantitativos (figura 3 del material adicional). El análisis inmunohistoquímico de la FLNC confirmó la ubicación intracelular de esta proteína en todas las muestras, pero en las muestras afectadas por FLNC-tv, esta proteína mostraba una distribución irregular y una ausencia completa en el disco intercalar (DI) (figura 5C). En cambio, en los tejidos afectados por FLNC-mRod2, estaba presente en el aparato contráctil, como en el grupo de control, pero especialmente positiva a nivel de los DI.
, que muestra el aumento del espacio intercelular en FLNC-tv y la compactación en FLNC-mRod2. B: hematoxilina de Heideinhain para las miofibrillas, que muestra la menor densidad en el caso de FLNC-mRod2. C: distribución intracelular de FLNC según lo determinado mediante inmunohistoquímica. Obsérvese la ausencia de FLNC en el disco intercalar de la muestra con FLNC-tv. D: identificación histoquímica de los colágenos fibrilares mediante tinción de picrosirio (en rojo) con un aumento de estas fibras en las muestras con afección genética. E y F: identificación inmunohistoquímica del colágeno tipo I (E) y tipo IV (F). Barra de escala: 20 μm. CTR: control; FLNC-mRod2: variante missense en el dominio FLNC-Rod2; FLNC-tv: variante truncada del gen de la filamina C.")
Patrón histológico, histoquímica miofibrilar, inmunohistoquímica de FLNC y análisis de remodelado de MEC en tejidos cardiacos con afección genética por FLNC. A: tinción de hematoxilina-eosina de muestras de control y de tejido cardiaco con afección genética (FLNC-tv y FLNC-mRod2), que muestra el aumento del espacio intercelular en FLNC-tv y la compactación en FLNC-mRod2. B: hematoxilina de Heideinhain para las miofibrillas, que muestra la menor densidad en el caso de FLNC-mRod2. C: distribución intracelular de FLNC según lo determinado mediante inmunohistoquímica. Obsérvese la ausencia de FLNC en el disco intercalar de la muestra con FLNC-tv. D: identificación histoquímica de los colágenos fibrilares mediante tinción de picrosirio (en rojo) con un aumento de estas fibras en las muestras con afección genética. E y F: identificación inmunohistoquímica del colágeno tipo I (E) y tipo IV (F). Barra de escala: 20 μm. CTR: control; FLNC-mRod2: variante missense en el dominio FLNC-Rod2; FLNC-tv: variante truncada del gen de la filamina C.
Por otro lado, el análisis de remodelado reveló la presencia de alteraciones fibrosas en las muestras con alteración genética en comparación con las de control (figura 5D), y mostró un claro aumento de las fibras de colágeno fibrilares. En las muestras afectadas por FLNC-mRod2, este aumento de las fibras de colágeno se observó alrededor de los miocardiocitos, que mostraban un patrón compactado y bien definido diferente del observado con la FLNC-tv (abundante y con una organización irregular) (figura 5E,F). Por último, el proceso fibrótico se confirmó de forma semicuantitativa, y se puso de manifiesto que el contenido de colágeno aumentado alcanzaba un 49,2±7,9% en las muestras afectadas genéticamente por FLNC-tv seguido de un 29±2,5% en las afectadas por FLNC-mRod2, en ambos casos considerablemente superiores al 26,5±2,2% de contenido de colágeno observado en el control (figura 3 del material adicional). Además, el remodelado de la red de colágeno (respuesta fibrosa) se acompañaba de una alteración de las fibras reticulares y del contenido de proteoglicanos ácidos, así como de su intensidad y su patrón (figura 4 del material adicional). Todas estas observaciones histológicas en las muestras afectadas por FLNC-mRod2 fueron uniformes en las 3 muestras.
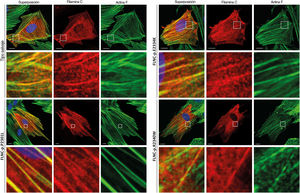
Análisis funcionalesSe realizó una transfección de células HT1080 y H9C2 con plásmidos de expresión para el FLNC de tipo salvaje y mutante (p.P2301L, p.E2334K y p.R2340W). Los análisis confocales revelaron una localización celular comparable para el FLNC de tipo salvaje y mutante en las células con transfección (figura 6; figura 5 del material adicional), lo cual indicaba que la agregación proteica patogénica era improbable con las variantes missense descritas.
. La filamina C marcada con c-Myc se muestra en rojo. La actina F (verde) se contuvo mediante faloidina conjugada con Alexa-488. Se muestran imágenes de células representativas. Barra de escala, 10μm.")
Se realizó una transfección de células H9C2 con plásmidos de expresión para la filamina C de tipo salvaje y mutante (p.P2301L, p.E2334K y p.R2340W). La filamina C marcada con c-Myc se muestra en rojo. La actina F (verde) se contuvo mediante faloidina conjugada con Alexa-488. Se muestran imágenes de células representativas. Barra de escala, 10μm.
Recientemente se han descrito esporádicas variantes missense de FLNC asociadas con la aparición de una MCR4–6. También se ha propuesto una asociación con la MCH7. No obstante, la evidencia procede de un pequeño número de publicaciones. Describimos aquí, por primera vez, un fenotipo cardiaco distintivo en una cohorte multicéntrica de pacientes con variantes missense raras en el dominio ROD2 del gen FLNC, que consisten en un fenotipo de MCH/MCR grave con una inusual HTVI en dientes de sierra. El solapamiento de MCH y MCR se ha descrito anteriormente en el contexto de mutaciones de los genes de proteínas sarcoméricas cardiacas21. Sin embargo, hasta la fecha se han publicado pocos casos de miocardio en dientes de sierra, y ninguno de ellos se ha asociado con un gen específico22. Tiene interés señalar que la mayoría de los pacientes presentaban el aspecto en dientes de sierra en las exploraciones de imagen cardiovasculares y que algunos lo mostraban a una edad temprana. Aunque el miocardio en dientes de sierra y la MCNC pueden considerarse epifenómenos en el contexto de otro trastorno, 3 publicaciones separadas con pacientes individuales han señalado un vínculo entre la MCNC y las mutaciones missense en FLNC1,23,24. Aunque los pacientes con el patrón en dientes de sierra mostraban recesos miocárdicos profundos, en todos los pacientes había una capa miocárdica única y compactada, en vez de 2 capas diferenciadas. En consecuencia, los criterios diagnósticos actuales de la MCNC no eran aplicables. Tiene interés señalar que Roldán-Sevilla et al.6 describieron a un individuo con la variante p.Pro2301Leu, como el paciente de la familia G, con una MCR en ausencia de HTVI. Aunque la descripción fenotípica es sucinta, esa diferencia con nuestro paciente puede explicarse por la ya conocida expresión pleotrópica de FLNC1.
Por lo que respecta a la repercusión clínica de las variantes FLNC-mRod2, la mayoría de los individuos tenían síntomas de IC y una notable disfunción diastólica. Es de destacar que, en esta pequeña cohorte de pacientes, en su mayoría jóvenes, hubo una gran incidencia de eventos clínicos mayores. Esto es coherente con una forma de miocardiopatía más grave que la MCH clásica y similar a la MCH debida a mutaciones en los genes de las proteínas filamentosas finas como la troponina T25. Esta observación podría revelar una fisiología restrictiva y una IC avanzada en las variantes FLNC-mRod2, que puede ser común en las fases iniciales de la enfermedad, de manera similar a las descripciones previas de pacientes con una IC avanzada a una edad temprana4,6. Es interesante que se observara una alta incidencia de variantes de novo, lo cual respalda la probabilidad de patogenicidad de estas variantes raras. Dado que se confirmó la paternidad en 4 variantes, estas se clasifican como probablemente patogénicas según los criterios del ACMG18.
Además, se obtuvo evidencia que indica una cosegregación fenotipo-genotipo leve en 4 familias no relacionadas, lo cual es un hecho crucial que respalda la patogenicidad. La FLNC es una proteína homodimérica codificada por el gen FLNC (7q32), que está formado por 47 exones codificadores. La proteína está formada por un dominio de unión de la actina aminoterminal y una ROD de 24 dominios de tipo Ig que están conectados por regiones bisagra flexibles entre los dominios 15 y 16 (bisagra 1) y los dominios 23 y 24 (bisagra 2)26. Sin embargo, se han descrito variantes de FLNC dispersas por todo el gen, y la información sobre genotipo-fenotipo y la afección cardiaca es incompleta; en estudios previos se ha cuestionado la gravedad clínica e incluso la patogenicidad de las variantes de FLNC frecuentes en las formas clásicas de MCH, que son casos claramente diferentes de los presentados en nuestra cohorte27: todos nuestros probandos eran portadores de una mutación missense situada en el mismo subdominio de ROD2, que afectaba concretamente a los dominios de tipo Ig del 18 al 21 (d18-21), y la mayor parte de ellas tenían en común este fenotipo singular de «miocardio en dientes de sierra». Los 2 pares de dominios (18-19 y 20-21) muestran conservación de todas las filaminas de los vertebrados28. Esta zona ROD2 contiene el dominio encargado de la unión de las glucoproteínas de membrana y forma una ROD semiflexible con 2 regiones bisagra y 24 repeticiones en tándem de alta homología, formadas por los residuos aminoácidos 93 a 103 organizados como dominios de tipo Ig. Los datos recientes sobre las correlaciones genotipo-fenotipo entre los portadores de variantes patogénicas de FLNC apuntan a un cluster de variantes missense en esta región y la aparición del fenotipo de MCH1. Este cluster d18-21 interactúa con las proteínas del disco Z, el desarrollo muscular y las proteínas relacionadas con la contracción. Además, tiene un especial interés, ya que es un punto crucial para la fosforilación proteica29. Se ha indicado que este subdominio ROD2 es esencial para la dimerización de la FLNC y la adquisición de la estructura proteica secundaria23. Así pues, las variantes missense en el subdominio ROD2 pueden producir una proteína con mal plegamiento y un deterioro de los enlaces cruzados y dar lugar a desorden sarcomérico y deterioro de la mecanotransducción23,30. Esta hipótesis deberá confirmarse en futuros estudios.
Se caracterizó por histología el tejido de FLNC-mRod2 y FLNC-tv en muestras cardiacas procedentes de 3 corazones explantados y de 1 caso de muerte súbita cardiaca respectivamente. Aunque debe interpretarse con precaución debido a la pequeñez de la muestra, los análisis histológicos e histoquímicos del paciente con FLNC-mRod2 mostraron células finas con una baja densidad de miofibrillas, lo cual puede explicarse por un deterioro de los enlaces cruzados de las proteínas estructurales, contráctiles o incluso citoesqueléticas de membrana de anclaje. Además, no se observaron agregaciones de FLNC en la histología ni en los experimentos funcionales. En este estudio, tal como se ha descrito anteriormente4,7, se utilizaron 2 sistemas heterólogos, y la línea celular H9C2 era más representativa de la fisiología miocítica. Aunque se ha observado una proteína anormal formando agregados en el tejido de pacientes con MCH y MCR asociadas a FLNC31, Valdes-Mas7 y Brodehl4 han demostrado que no todas las variantes missense de FLNC asociadas con miocardiopatía inducen una agregación proteica, por ejemplo en el caso de las variantes de FLNC-mRod2 p.I2160F o p.V2297M4,5. Es interesante que la ausencia de agregados se ha descrito en el tejido cardiaco de pacientes con FLNC-tv en el dominio ROD2, lo cual indica la ausencia de una proteína FLNC anormal23.
También se confirmó la localización intracelular de la FLNC en todas las muestras cardiacas, pero hubo diferencias entre ellas. Mientras que en el caso de FLNC-mRod2 había abundancia de FLNC en los DI, las muestras afectadas por FLNC-tv expresaban una proteína de distribución irregular con una ausencia completa en los DI. Esta ausencia de FLNC en el DI en la FLNC-tv se había descrito anteriormente en modelos de pez cebra31. Además, se confirmó un remodelado de la MEC diferente, dependiente del genotipo, que conducía a alteraciones fibróticas diversas en los pacientes con una afección genética. Los tejidos afectados por FLNC-tv estaban ocupados por una red de colágeno fibrilar abundante y de distribución irregular, lo cual confirma una respuesta fibrosa evidente9. Sin embargo, las muestras afectadas por FLNC-mRod2 presentaban un patrón de tejido bien definido y compactado, con un proceso fibroso de leve a moderado. Esto contrasta con los resultados del realce tardío de gadolinio, pero puede explicarse por la fibrosis miocárdica difusa que no puede evaluarse mediante las imágenes de realce tardío de gadolinio. Los futuros estudios con mapeo T1 permitirán mejorar la evaluación de la fibrosis miocárdica intersticial.
LimitacionesLas principales limitaciones de este estudio son el pequeño número de pacientes evaluados y su diseño retrospectivo. De las 9 variantes raras, 5 se consideran variantes de significado desconocido, lo cual limita las conclusiones que pueden extraerse. Por lo que respecta a la histología, dada la naturaleza de las muestras, se incluyeron solo unas pocas muestras cardiacas independientes afectadas por este trastorno genético concreto, y ello limita la posibilidad de realizar análisis cuantitativos y estadísticos. La caracterización del tejido en la resonancia magnética cardiaca tiene la limitación del carácter retrospectivo del estudio. Además, es posible que las células HT1080 no reproduzcan la fisiología de los miocardiocitos humanos.
CONCLUSIONESLas raras variantes missense de FLNC en el dominio ROD2 pueden mostrar un fenotipo solapado que comprende la MCH y la MCR, con una HTVI en dientes de sierra caracterizada por una progresión grave de la IC y un remodelado histopatológico cardiaco distintivo (figura 7).

Ilustración central. Identificación y manifestaciones clínicas de los pacientes con variantes missense en el dominio ROD2 del FLNC y miocardio en dientes de sierra. Se incluyó a pacientes evaluados genéticamente a causa de una miocardiopatía hipertrófica/miocardiopatía restrictiva que presentaban una variante missense en el dominio ROD2 del gen FLNC: 11 probandos mostraron un miocardio en dientes de sierra: el cribado en cascada identificó hasta 15 individuos con ese rasgo; en 6 familias se observó una cosegregación leve. Los pacientes contrajeron una insuficiencia cardiaca a una edad temprana. El examen anatomopatológico mostró un remodelado cardiaco bien definido. FLNC: filamina C; MCH: miocardiopatía hipertrófica; MCR, miocardiopatía restrictiva; NYHA: calse funcional de la New York Heart Association.
A. Brodehl agradece el apoyo económico de la Ruhr-University Bochum (FoRUM, FoRUM-F937R2-2020). F.J. Bermúdez Jiménez recibió apoyo económico de un programa de formación en investigación Río Hortega del Instituto de Salud Carlos III (CM19/00227).
CONTRIBUCIÓN DE LOS AUTORESTodos los autores hicieron una contribución significativa a este trabajo reclutando los casos en sus centros de referencia. F.J. Bermúdez-Jiménez y V. Carriel redactaron y coordinaron por igual el manuscrito. A.B. llevó a cabo el estudio funcional. J. Jiménez-Jáimez supervisó el trabajo clínico y de investigación y la versión final del manuscrito. Todos los autores han aprobado la versión final del manuscrito.
CONFLICTOS DE INTERESESNinguno.
- –
La FLNC es una proteína del músculo esquelético codificada por el gen FLNC, que actúa como proteína de señalización y de estructura.
- –
Se ha observado una intensa asociación de las variantes del gen FLNC con la miocardiopatía dilatada arritmógena y esporádicamente con la miocardiopatía hipertrófica, la miocardiopatía restrictiva y la miocardiopatía no compactada.
- –
Este estudio proporciona una nueva perspectiva sobre el espectro de la miocardiopatía asociada con FLNC y describe, por primera vez, una asociación novedosa con un fenotipo cardiaco solapado, formado por el miocardio en dientes de sierra y la miocardiopatía hipertrófica y restrictiva.
- –
Este es el primer estudio en el que se asocia un fundamento genético con el patrón de miocardio en dientes de sierra.
- –
Deberá evaluarse el gen FLNC en pacientes con este fenotipo cardiaco, inespecífico pero grave, para determinar el diagnóstico apropiado e identificar a los familiares en riesgo.
Quisiéramos dar las gracias a los pacientes y a sus familias por su generosa e incondicional colaboración. Damos las gracias a Fabiola Bermejo Casares, Paloma de la Cueva Batanero (Departamento de Histología, Universidad de Granada, España), Caroline Stanasiuk (EHKI, Alemania), Raúl Franco Gutiérrez (Hospital Universitario Lucus Augusti, Santiago de Compostela, España), Neus Baena Díez (Hospital Universitario Parc Taulí, Sabadell, España), Carlos Gómez Navarro (Hospital Universitario Torrecárdenas, Almería, España) y Huafrin Kotwal (St. Bartholomew's Hospital, Londres, Reino Unido) por la asistencia técnica. Los análisis histológicos contaron con el apoyo del Tissue Engineering Group (CTS-115), Universidad de Granada, España.