
Las glucogenosis pueden asemejarse a la miocardiopatía hipertrófica (MCH)1. Algunas de las glucogenosis identificadas más recientemente, también con afección cardiaca, son prácticamente desconocidas y, por lo tanto, es difícil sospechar su presencia (tabla 1). En la presente comunicación se destaca que la glucogenosis tipo XV (OMIM 613507) también puede presentarse como una miocarditis o incluso suscitar el diagnóstico de miocardiopatía arritmogénica del ventrículo izquierdo (VI).
Resumen de las glucogenosis con afectación cardíaca
| Enfermedad, año de descubrimiento | Déficit enzimático | Gen | Patrón hereditario | Acumulación de poliglucosanos | Órganos clínicamente afectados (otros órganos con depósitos) | Información adicional |
|---|---|---|---|---|---|---|
| Glucogenosis II, Pompe, 1932 | Maltasa ácida | GAA | Autosómico recesivo | No | MÚSCULO, corazón, arterias | Hipotonía, infecciones respiratorias, retraso en el desarrollo, intervalo PR corto, síndrome de Wolff-Parkinson-White, bloqueo auriculoventricular, miocardiopatía hipertrófica, lengua e hígado agrandados, aneurismas |
| Glucogenosis IIB, Danon, 1981 | Proteína 2 de la membrana relacionada con lisosomas | LAMP2 | Dominante ligado al cromosoma X | No | CORAZÓN, músculo | Intervalo PR corto, síndrome de Wolff-Parkinson-White, miocardiopatía hipertrófica, deterioro cognitivo, miopatía, transaminasas elevadas, alteraciones visuales. Los varones suelen verse más afectados que las mujeres |
| Glucogenosis IIIa, Cori-Forbes, 1952 | Enzima desramificadora (amilo-1,6-glucosidasa) | AGL | Autosómico recesivo | No | HÍGADO, corazón, afección multisistémica | Hepatomegalia, hipertransaminasemia, cirrosis, adenomas hepáticos, hepatocarcinoma, hipoglucemia, creatincinasa elevada, miocardiopatía hipertrófica, estatura baja, acidosis láctica, dislipemia, hipogonadismo, anomalías faciales y otitis media |
| Glucogenosis IV, Andersen, 1956 | Enzima ramificante (amilo 1,4-1,6 transglucosidasa) | GBE1 | Autosómico recesivo | Sí | MÚSCULO, corazón, hígado, cerebro | Miopatía con o sin afección cardíaca (miocardiopatía hipertrófica, miocardiopatía dilatada), neuropatía y cirrosis |
| Glucogenosis XIV, 2007 | Fosfoglucomutasa | PGM1 | Autosómico recesivo | No | Corazón, músculo, multisistémico | Miocardiopatía dilatada, fisura palatina, úvula bífida, fenotipo de Pierre Robin, rabdomiolisis, hipertermia maligna, insuficiencia suprarrenal, hipoglucemia, aumento de la transaminasemia, hipogonadismo, retraso del crecimiento, alteraciones de la coagulación. Es posible que la biopsia muscular no detecte la acumulación de glucógeno |
| Glucogenosis XV, 2010 | Glucogenina 1 | GYG1 | Autosómico recesivo | Sí | CORAZÓN, músculo | La glucogenosis XV se refiere a la afección cardíaca (miocardiopatía hipertrófica, miocardiopatía dilatada) en esta enfermedad, mientras que la enfermedad muscular se conoce como miopatía por cuerpos de poliglucosano de tipo 2 (es muy infrecuente que ambas afecciones coexistan) |
| Miocardiopatía hipertrófica VI (MCH VI), 1966-1985 | Subunidad gamma de la proteincinasa activada por AMP | PRKAG2 | Autosómico dominante | Sí | CORAZÓN, músculo | Los pacientes pueden tener también miocardiopatía hipertrófica masiva, síndrome de Wolff-Parkinson-White y bloqueo auriculoventricular (a menudo, solo se observa afección cardíaca) |
| Inmunodeficiencia, autoinflamación, miocardiopatía (miopatía por cuerpos de poliglucosano de tipo 1), 2009 | Ligasa E3 de la ubiquitina | RBCK1 | Autosómico recesivo | Sí | CORAZÓN, MÚSCULO, hígado | Miocardiopatía dilatada, miopatía y septicemia |
Órgano afectado predominantemente, en mayúsculas.
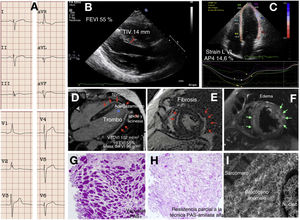
Un probando joven refirió dolor torácico limitante, episodios presincopales mal definidos y debilidad progresiva. Se detectó un ligero aumento de la concentración sanguínea de troponina T y de la concentración de proteínas en la orina, con la creatincinasa normal. El electrocardiograma demostró ritmo sinusal, bloqueo completo de la rama derecha del haz de His atípico con hemibloqueo posterior izquierdo (figura 1A); se observaron extrasístoles ventriculares y supraventriculares infrecuentes y aislados mediante Holter y registrador implantable. La angiografía coronaria por tomografía computarizada descartó estenosis coronarias. La imagen cardiaca mediante ecocardiografía y resonancia magnética cardiaca identificó anomalías estructurales en el VI. Las alteraciones consistieron en hipertrofia de leve a moderada del VI solo en el segmento basal del tabique, fracción de eyección del VI (FEVI) en el límite inferior, strain longitudinal global ligeramente reducido, volumen ligeramente incrementado, paredes apical y lateral adelgazadas e hipocinéticas con un trombo laminar, edema epicárdico o intramiocárdico con fibrosis asociada grave (figura 1B-F). El diagnóstico fue de miocarditis y inició tratamiento con colchicina, bisoprolol y anticoagulantes orales. El paciente sufrió un ictus cerebeloso poco tiempo después con recuperación ad integrum pero, por lo demás, los síntomas y las pruebas no han variado con un seguimiento de 24 meses. El dolor torácico persistente motivó la realización de una biopsia endomiocárdica, en la que se vio que no había confusión o disarray ni fibrosis relevante pero sí vacuolación miocárdica grave que desplazaba periféricamente los núcleos. La positividad marcada del ácido peryódico de Schiff (PAS) en las vacuolas dio el diagnóstico de glucogenosis, y su atenuación parcial si se incluía tratamiento previo con diastasa en el protocolo de tinción, fue consistente con depósitos de poliglucosano (figura 1G,H). Las imágenes ultraestructurales también concordaron con el diagnóstico (figura 1I). Se evaluaron los genes de glucogenosis, MCH y miocardiopatía arritmogénica mediante secuenciación de nueva generación (NextSeq 500, Illumina Technologies). El probando era portador de 2 variantes patogénicas ya publicadas del gen GYG-1, a saber: p.Asp102His y p.Gly135Arg2. La exploración clínica exhaustiva por un neurólogo experto, la tomografía computarizada de todo el cuerpo, las pruebas estándar para evaluar la función respiratoria y una prueba de esfuerzo permitieron descartar la presencia de afectación de la musculatura esquelética. La necesidad de mantener la anticoagulación impidió la realización de una biopsia muscular. Con respecto al tratamiento, se eliminaron la colchicina y los bloqueadores beta y se intensificaron los anticoagulantes. Los 6 sujetos del estudio familiar de 3 generaciones realizado resultaron negativos para el fenotipo y solo eran heterocigotos para una de las mutaciones.
, el asterisco resalta el trombo apical y las flechas verdes señalan el edema miocárdico. G y H: histología. I: caracterización ultraestructural. FEVI: fracción de eyección del ventrículo izquierdo; TIV: grosor del tabique interventricular; PAS: reacción del ácido peryódico de Schiff; Strain LVI AP4: strain longitudinal del VI en el plano apical de 4 cámaras; VI: ventrículo izquierdo; VTDVI: volumen telediastólico del VI. Esta figura se muestra a todo color solo en la versión electrónica del artículo.")
Fenotipo cardiaco de la glucogenosis tipo XV. A: electrocardiograma. B y C: ecocardiografía. D-F: imágenes de resonancia magnética cardiaca; las flechas rojas señalan el realce tardío de gadolinio (fibrosis), el asterisco resalta el trombo apical y las flechas verdes señalan el edema miocárdico. G y H: histología. I: caracterización ultraestructural. FEVI: fracción de eyección del ventrículo izquierdo; TIV: grosor del tabique interventricular; PAS: reacción del ácido peryódico de Schiff; Strain LVI AP4: strain longitudinal del VI en el plano apical de 4 cámaras; VI: ventrículo izquierdo; VTDVI: volumen telediastólico del VI. Esta figura se muestra a todo color solo en la versión electrónica del artículo.
En la literatura se prefiere el término glucogenosis tipo XV para hacer referencia al fenotipo miocardiopático, mientras que el nombre de miopatía por cuerpos de poliglucosano de tipo 2 (PGBM2, OMIM 616199) se aplica a una enfermedad alélica que se caracteriza por una miopatía de evolución lenta e inicio tardío sin afectación cardiaca2–5. Hasta el momento, se han descrito 38 pacientes con PGBM2, y el del presente estudio es el quinto caso de glucogenosis tipo XV2–4. Todos estos pacientes presentan 2 mutaciones del gen GYG-1 y se identifica acumulación de poliglucosanos tanto en los cardiomiocitos como en el músculo esquelético. Destaca que las mutaciones sin sentido son más frecuentes en los pacientes con miopatía que en aquellos con miocardiopatía3,5.
Después de revisar la literatura3,4 y a nuestro probando, destacamos que la glucogenosis tipo XV representa un diagnóstico cardiológico, con afección biventricular microscópica, aunque las técnicas de imagen cardiaca solo detectan una enfermedad aislada del VI. Cabe destacar que en esta enfermedad no necesariamente se observa hipertrofia masiva del VI (14-23 mm en los 5 pacientes identificados hasta ahora) y nunca se ha relacionado con deterioro cognitivo, bloqueo auriculoventricular o preexcitación, como sí ocurre en otras glucogenosis conocidas, como las enfermedades PRKAG2, de Danon y de Pompe1. En cambio, el dolor torácico de larga duración (80%), las alteraciones regionales del VI (adelgazamiento, 60%; hipocinesia o acinesia, 40%), las alteraciones de la caracterización tisular del VI (cicatriz, 100%; edema, 20%) y los episodios de embolia sistémica (ictus, 40%; trombosis del VI, 20%) son los rasgos más representativos. Además, la disfunción del VI es muy variable (FEVI 25-55%, volúmenes de ligera a gravemente aumentados y necesidad de trasplante cardiaco, 20%), se asocia con taquiarritmias ventriculares potencialmente mortales (40%) y raramente con miopatía esquelética (20%). Curiosamente, la enfermedad es muy penetrante en varones pero no en mujeres (la única mujer reportada, homocigota para la mutación p.Asp102His, no mostró ningún signo o síntoma en la evaluación cardíaca y de músculo esquelético2).
Por último, cabe destacar que la glucogenosis tipo XV debería considerarse una fenocopia no solo de MCH, sino también de miocarditis y de miocardiopatía arritmogénica dominante del VI (sobre todo si la hipertrofia del VI es únicamente leve y la cicatrización, pronunciada). La biopsia endomiocárdica y los estudios histológicos son cruciales para dirigir la evaluación genética. En caso de sospecha, los cardiólogos deberían asegurarse de incluir el GYG-1 en las pruebas seleccionadas para el estudio genético. Lamentablemente, en el momento actual no hay ningún tratamiento para esta enfermedad. El tratamiento de las arritmias y la insuficiencia cardiaca es sintomático.
FINANCIACIÓNEste estudio fue financiado parcialmente por subvenciones del Instituto de Salud Carlos III, el FEDER de la Unión Europea, Una forma de hacer Europa [PI18/01582] y el Biobanco La Fe [PT17/0015/0043], y el Memorial Nacho Barberá.