
Se presenta el caso de un niño de 9 años, sin antecedentes personales ni familiares de enfermedades cardiovasculares, que sufrió una parada cardiaca extrahospitalaria mientras jugaba al fútbol en el colegio. Después de 7min de maniobras de reanimación avanzada, el paciente recuperó la circulación espontánea.
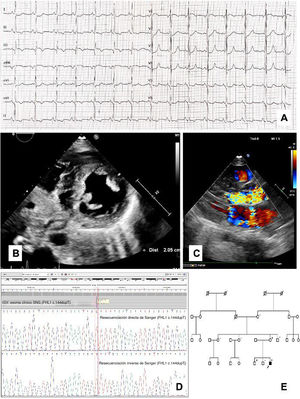
Al ingreso en la unidad de cuidados intensivos pediátricos, el electrocardiograma (ECG) del paciente mostraba ritmo sinusal, ondas T aplanadas y depresión del segmento ST en las derivaciones inferolaterales y ondas Q patológicas en aVR (figura 1A). La ecocardiografía mostró hipertrofia del ventrículo izquierdo (HVI) asimétrica, con tabique interventricular de 21mm (Z score > +6), función sistólica conservada y movimiento anterior sistólico de la válvula mitral, lo que motivaba un gradiente en el tracto de salida del ventrículo izquierdo (TSVI) de hasta 90mmHg, con posterior insuficiencia mitral moderada (figuras 1B y C). La exploración neurológica fue normal, sin afección de los músculos o las articulaciones de las extremidades y sin aumento anómalo de la creatincinasa. Doce días después del ingreso, se implantó un desfibrilador automático (DAI) subcutáneo en prevención secundaria. El paciente tuvo una recuperación satisfactoria, comenzó tratamiento con propranolol y permaneció asintomático hasta 1 año después, cuando tuvo una descarga apropiada del DAI.

A: electrocardiograma del caso inicial en diagnóstico que muestra repolarización anómala en derivaciones inferolaterales y ondas Q patológicas en aVR. B: ecocardiografía bidimensional en la proyección paraesternal de eje corto de los músculos papilares, que muestra HVI asimétrica. C: vista paraesternal de eje largo de la ecocardiografía Doppler de flujo de color transtorácico, que muestra obstrucción del tracto de salida del ventrículo izquierdo e insuficiencia moderada de la válvula mitral posterior. D: detección y validación de la variante c.144dupT de FHL1 en Integrative Genomics Viewer y Chromas-Sanger Viewer. E: genealogía familiar; la flecha indica el caso índice. IGV: Integrative Genomics Viewer; SNG: secuenciación de nueva generación.
Se recogió una muestra de sangre para el análisis de exoma clínico mediante secuenciación de nueva generación (NGS) que incluyó 199 genes relacionados con cardiopatía. Se identificó una nueva variante nonsense en el exón 2 del gen FHL1 (cromosoma X) en hemicigosis: NM_001159699: c.144dupT;p. (Asp49*), que se consideró probablemente patógena (figura 1D). No se encontraron otras variantes relevantes en genes asociados con miocardiopatía hipertrófica (MCH).
Se hicieron pruebas genéticas y clínicas a la madre y los 2 hermanos del paciente. Se demostró que la madre era portadora de la variante, con fenotipo normal. Los hermanos no tenían un fenotipo ecocardiográfico con alteraciones, aunque el ECG del hermano mayor mostró anomalías sutiles, como ondas Q patológicas y ondas T aplanadas en las derivaciones laterales. El resultado del estudio genético de cosegregación fue negativo en el hermano menor, pero la variante sí estaba presente en el hermano mayor. También se hizo la prueba a la abuela, las tías y el tío maternos; todos presentaron un fenotipo normal y ninguno era portador de la mutación (figura 1E). No se pudo hacer la prueba al abuelo materno del caso índice porque ya había fallecido. En este escenario, ni los tíos ni la abuela eran portadores de FHL1 c.144dupT, por lo que se supone que la variante sería una mutación de novo en la madre.
Se describe el caso de un niño con MCH, cuya presentación fue un episodio de muerte súbita recuperada, portador de una nueva variante no descrita antes y probablemente patogénica en un gen no sarcomérico con patrón de herencia ligada al cromosoma X. Se ha descrito que FHL1 es un gen patogénico implicado en la etiología de varias miopatías esqueléticas, a veces junto con miocardiopatía dilatada, no compactada o, con más frecuencia, hipertrófica1,2. Se encuentra en el cromosoma Xq26.3 y codifica el dominio four and a half LIM domains protein 1 (LIM 4.5 1), un miembro de la familia de proteínas LIM. Cada dominio LIM contiene 2 dedos de zinc de unión, que están muy conservados. Las mutaciones que afectan a estas regiones suelen ser patogénicas2,3. FHL1 es la principal isoforma en el músculo esquelético y cardiaco y desempeña un papel importante en la síntesis de proteínas sarcoméricas, la integridad y la señalización intracelulares, la transcripción génica y la detección de estrés biomecánico2.
Además, cada vez hay más pruebas de que algunas mutaciones en FHL1 también podrían explicar la patogénesis de casos atípicos de MCH aislada que no se puede explicar por mutaciones sarcoméricas tradicionales. FHL1 es el único miembro de la familia de proteínas LIM con diferentes variantes de empalme (splicing) y sus mutaciones generan péptidos dañinos que causan hipertrofia cardiaca, disfunción diastólica o deterioro de la contractilidad miocárdica1,4.
La tabla 1 resume las publicaciones previas de los pocos casos y familias con MCH sin miopatía que se han relacionado con variantes de FHL1 (en heterocigosis o hemicigosis). El patrón de HVI es heterogéneo y hay algunos casos notificados de anatomía «esponjosa» que no cumplen los criterios de VI no compactado4,5. Los varones portadores de variantes truncadas tienden a presentar un curso clínico muy agresivo y precoz tanto de la insuficiencia cardiaca avanzada como de las arritmias ventriculares malignas. En cambio, las mujeres heterocigotas descritas hasta la fecha presentan una afección cardiaca más leve1.
Casos descritos anteriormente de MCH aislada debida a mutaciones de FHL1
| Sexo y edad en el momento del diagnóstico | Variante de FHL1 | Fenotipo cardiaco | Eventos cardiovasculares | Referencia bibliográfica |
|---|---|---|---|---|
| Varón, 16 años | c.134delA, p.Ser45fsNonsense | MCH, predominio septal. Obstrucción del TSVI. TIV 20mm. Alteraciones del ECG | Friedrich et al. Hum Mol Genet. 20122 | |
| Mujer, 43 años | c.459C>A, p.Cys153XNonsense | MCH. TIV 17mm. Alteraciones del ECG | Friedrich et al. Hum Mol Genet. 20122 | |
| Varón, 18 años | c.599_6000insT, p.F2000fs32XNonsense | MCH aislada, restrictivo. TIV 24mm. Insuficiencia mitral moderada. Alteraciones del ECG | Trasplante de corazón a los 31 años (IC en fase terminal) | Hartmannova et al. Circ Cardiovasc Genet. 20131 |
| Varón, 55 años | c.599_6000insT, p.F2000fs32XNonsense | MCH aislada. TIV 20mm. Insuficiencia mitral moderada. Alteraciones del ECG | Trasplante de corazón a los 59 años (IC en fase terminal) | Hartmannova et al. Circ Cardiovasc Genet. 20131 |
| Varón, 38 años | c.468_469insC.NM_001159700_1Nonsense | MCH, predominio apical y medioventricular. TIV 17mm. Dilatación biauricular. Disfunción diastólica. FEVI 56%. RM con realce tardío de gadolinio | Fibrilación auricularMuerte a los 61 años (fibrosis pulmonar, insuficiencia pulmonar y multiorgánica) | Gallego-Delgado et al. Int J Cardiol. 20153 |
| Mujer, 32 años | c.468_469insC.NM_001159700_1Nonsense | MCH, predominio apical y medioventricular. TIV 21mm. Disfunción diastólica. FEVI 73%. RM con realce tardío de gadolinio | Fibrilación auricularIctus a los 46 años | Gallego-Delgado et al. Int J Cardiol. 20153 |
| Varón, 8 años | c.468_469insC.NM_001159700_1Nonsense | MCH asimétrica. TIV 27mm. Obstrucción del TSVI. VI restrictivo. FEVI 23%. | MSC (TV) recuperada a los 8 años. Trasplante de corazón a los 17 años | Gallego-Delgado et al. Int J Cardiol. 20153 |
| Varón, 7 años | c.468_469insC.NM_001159700_1Nonsense | MCH obstructiva asimétrica. TIV 19mm. Insuficiencia mitral moderada | Prevención primaria con DAI a los 11 años. Descarga apropiada a los 17 años. MSC (TV, con una FC más baja que el umbral de DAI) a los 17 años | Gallego-Delgado et al. Int J Cardiol. 20153 |
| Varón, 19 años | c.267C>A, p.Cys89TerNonsense | MCH con fibrosis intersticial extensa en la autopsia. TIV 20mm | Parada cardiaca súbita | Gaertner-Rommel et al. Mol Genet Genomic Med. 20195 |
DAI: desfibrilador automático implantable; ECG: electrocardiograma; FC: frecuencia cardiaca; FEVI: fracción de eyección del ventrículo izquierdo; IC: insuficiencia cardiaca; MCH: miocardiopatía hipertrófica; MSC: muerte súbita cardiaca; RM: resonancia magnética; TIV: tabique interventricular; TSVI: tracto de salida del ventrículo izquierdo; TV: taquicardia ventricular; VI: ventrículo izquierdo.
La variante de [0] FHL1 c.144dupT en pacientes aún no estaba descrita ni se halla en las bases de datos poblacionales. Al igual que otras variantes patogénicas/probablemente patogénicas en FHL1, la mutación genera una proteína truncada. En este caso concreto, se predice que la variante generará una proteína acortada (49 aminoácidos en lugar de 296, con la pérdida de todos los elementos funcionales). Se considera probablemente patogénica según los criterios modificados del American College of Medical Genetics and Genomics (ACMG)6. Otro paciente con MCH aislada, notificado en 2012, mostró una mutación con una localización genómica muy cercana, c.134delA (p.Ser45fs)2.
El caso índice presentado sufrió una parada cardiaca a la edad de 9 años. Según la información disponible, solo se había publicado un caso semejante3. Los autores describen una familia con una nueva variante por desplazamiento del marco de lectura en FHL1 y MCH aislada, diagnosticada en un niño de 8 años que había sufrido una taquicardia ventricular. En cuanto al resto de la familia, la madre tenía un fenotipo normal en la primera evaluación. Este hallazgo está en consonancia con casos previos notificados de mujeres con fenotipo leve y similar a otras enfermedades con un patrón de herencia ligado al cromosoma X. El hermano mayor era portador de la variante, aunque no tenía un fenotipo claro cuando lo examinaron por primera vez. Sin embargo, la penetrancia incompleta y la expresividad variable son frecuentes en la MCH, así como en otras miocardiopatías. Cabe destacar que el paciente todavía es muy joven, de modo que está justificado un estrecho seguimiento.
La muerte súbita cardiaca (MSC) como presentación de la MCH es muy poco frecuente en la población pediátrica, pero potencialmente grave. En la actualidad, muchas investigaciones están tratando de determinar los principales factores de riesgo que deben tenerse en cuenta para evitar las arritmias ventriculares malignas en niños con MCH. Las pruebas genéticas son muy útiles para confirmar el diagnóstico, el cribado familiar y el asesoramiento genético. Sin embargo, con pocas excepciones y casos de doble mutación, la mayoría de las variantes causales de MCH no ofrecen información pronóstica sobre el riesgo arrítmico. Las pruebas genéticas disponibles actualmente, con grandes paneles de genes o exomas, pueden ofrecer más información sobre la estratificación del riesgo de MSC, aunque los resultados deben interpretarse con cautela.
Sin embargo, respecto a la MCH relacionada con FHL1, se cree que las pruebas publicadas y el caso que se presenta aquí demuestran que los pacientes varones jóvenes con MCH aislada y variantes truncadas en FHL1 parecen presentar un curso agresivo. Por lo tanto, para los varones portadores de una variante probablemente patogénica en FHL1 con MCH podría considerarse el implante precoz de un DAI.
Los autores aceptan toda la responsabilidad por el contenido del original según lo definido por el International Committee of Medical Journal Editors. La investigación comunicada se llevó a cabo de acuerdo con las recomendaciones internacionalmente aceptadas para la investigación clínica (Declaración de Helsinki de la World Medical Association, revisada en octubre de 2013). Los autores confirman que obtuvieron el consentimiento informado por escrito de la madre del paciente para su publicación en Rev Esp Cardiol y su difusión de forma impresa y electrónica.
FINANCIACIÓNLos autores no recibieron ningún ayuda económica.
CONTRIBUCIÓN DE LOS AUTORESM. López Blázquez: concepto y diseño del artículo, obtención de datos, redacción, aprobación final de la versión para publicación, garantía de la exactitud e integridad del artículo. A.I. Fernández Ávila: análisis e interpretación de los datos, aprobación final de la versión para publicación. R. Álvarez García-Rovés: obtención de datos. M. Centeno Jiménez: obtención de datos. C. Gómez González: revisión del contenido del artículo. M.Á. Espinosa Castro: concepto y diseño del artículo, revisión del contenido, aprobación final de la versión para publicación.
CONFLICTO DE INTERESESLos autores declaran que no hay ningún conflicto de intereses.