TPM1 es uno de los principales genes en la miocardiopatía hipertrófica (MCH). La información clínica sobre portadores es relativamente escasa, lo cual limita la interpretación de los estudios genéticos. Nuestro objetivo es establecer la correlación genotipo-fenotipo de la variante p.Arg21Leu de TPM1 en una serie de familias.
MétodosSe evaluó el TPM1 mediante secuenciación de nueva generación en 10.561 probandos con cardiopatías hereditarias. Se genotipificó a los familiares mediante Sanger. Se analizaron la cosegregación, las características clínicas y los eventos cardiovasculares. Se estimó la distribuición geográfica de las familias en Portugal y España.
ResultadosSe identificó la variente p.Arg21Leu de TPM1 en 25/4.099 (0,61%) casos con MCH y estaba ausente en 6.462 controles con otras cardiopatías familiares (p<0,0001). Se identificó a 83 portadores (31 probandos). La LOD score combinada para cosegregación fue 3,95. La probabilidad acumulada de diagnóstico en portadores a los 50 años fue del 50% para los varones y el 25% para las mujeres. El 17 de los varones y el 46% de las mujeres no estaban afectadas a los 70 años. El grosor medio del ventrículo izquierdo fue 21,4 ±7,65mm. El riesgo de muerte súbita-MCH fue bajo en 34 (77,5%), intermedio en 8 (18%) y alto en 2 (4,5%) de los portadores. La supervivencia libre de eventos cardiovasculares fue del 87,5% a los 50 años. El 6% de los portadores eran homocigotos y el 18% tenían una variante adicional. El origen de las familias se concentró en Galicia, Extremadura y norte de Portugal, lo que indica un efecto fundador.
ConclusionesP.Arg21Leu es una variante patogénica de TPM1 asociada con MCH de penetrancia tardía/incompleta y pronóstico generalmente favorable
Palabras clave
La miocardiopatía hipertrófica (MCH) es un trastorno genético frecuente (prevalencia > 1/500), con amplia heterogeneidad fenotípica y de locus1–3. Se considera al gen TPM1 (que codifica la alfatropomiosina) uno de los principales genes causantes de la MCH; sin embargo, es una etiología relativamente rara, que representa del 1 al 5% de los casos con este fenotipo3.
La información clínica disponible sobre la mayoría de los pacientes portadores de variantes de TPM1 se restringe a una sola familia o unos pocos probandos por cada variante4–13, lo que limita una interpretación clínica asertiva de los hallazgos genéticos. La excepción a esta regla es la variante p.Asp175Asn de TPM1, identificada en varios pacientes con MCH finlandeses, porque es el único efecto fundador en este gen descrito hasta la fecha14,15. Los estudios de correlación genotipo-fenotipo de variantes fundadoras en poblaciones con MCH han contribuido a una mejor comprensión del curso clínico y el pronóstico asociado con esta variante16–22.
En nuestro centro, se identificó la variante p.Arg21Leu de TPM1 mediante secuenciación de nueva generación en varios individuos con MCH, como los portadores homocigotos. Aunque se habían realizado estudios genéticos en varios países, los casos de la variante p.Arg21Leu procedían predominantemente de hospitales del noroeste de España y Portugal, lo que plantea la hipótesis de que podría ser un efecto fundador. Hasta la fecha, esta variante se ha clasificado en bases de datos públicas como de significado clínico incierto (5 suscriptores independientes) y probablemente patógena (solo 1 suscriptor)23, mientras que aún se desconoce el fenotipo asociado. El principal objetivo de este estudio es describir las características fenotípicas y el pronóstico asociado de la variante p.Arg21Leu de TPM1 en una serie de familias.
MÉTODOSSe trata de un estudio descriptivo de una serie de familias portadoras de la variante NP_001018005.1: p.Arg21Leu de TPM1 (c.62G >T). Este estudio se llevó a cabo de acuerdo con los principios de la Declaración de Helsinki y forma parte de la línea de investigación registrada con el número 2012/139 en el Comité de Ética de la Investigación de Galicia, España. Se obtuvo el consentimiento informado de todos los participantes.
Población de estudio y análisis bioinformáticoDesde marzo de 2008 hasta septiembre de 2018, se secuenció el gen TPM1 en 10.561 probandos consecutivos con diferentes cardiopatías hereditarias de distintos hospitales de todo el mundo, remitidos a nuestro centro para que se estableciera el diagnóstico molecular.
Todos los exones y las regiones intrónicas adyacentes de 213 genes (tabla 1 del material adicional) relacionados con miocardiopatías y muerte súbita cardiaca (MSC) se estudiaron mediante secuenciación de nueva generación en los probandos. La profundidad mínima de lectura obtenida fue > 30 (promedio de 250× a 400×) en ácido desoxirribonucleico (ADN). Los fragmentos que no cumplían estos criterios se secuenciaron mediante Sanger. La genotipificación en cascada familiar de parientes se realizó mediante secuenciación de Sanger. Un equipo multidisciplinario realizó el análisis bioinformático y la interpretación clínica. La información sobre la frecuencia alélica de la población general (controles) se consideró en función de las poblaciones de la base de datos Genome Aggregation Database (gnomAD) y del programa TOPMed. La clasificación de la patogenicidad de las variantes fue conforme a las recomendaciones actuales, que tienen en cuenta la prevalencia de la variante en individuos afectados frente a los controles, datos de cosegregación familiar, estudios funcionales y predictores in silico, entre otros criterios24. Se describieron variantes patogénicas/posiblemente patogénicas en cualquiera de los genes secuenciados, así como aquellas de significado clínico incierto en 18 genes de la MCH prioritarios (tabla 1 del material adicional).
Además, se establecieron contactos durante el primer encuentro ibérico sobre miocardiopatías (Óbidos, Portugal, marzo de 2017) para recopilar información de portadores de p.Arg21Leu de TPM1 identificados en otros centros portugueses y españoles.
Caracterización fenotípicaSe revisaron los datos clínicos de los probandos y familiares, y se realizaron los árboles genealógicos. Se incluyó a todas las familias portadoras, independientemente de la duración del seguimiento o del número de familiares evaluados. Se analizaron los factores relacionados con la penetrancia y la expresión de la enfermedad en la MCH: edad, sexo, genotipo, características clínicas y factores de riesgo de MSC. El inicio temprano se definió como diagnóstico en personas menores de 35 años25. Los criterios diagnósticos y el modelo de estratificación del riesgo de MSC en MCH siguieron las recomendaciones de la Sociedad Europea de Cardiología (ESC)3. Todos los probandos cumplían los criterios diagnósticos convencionales de MCH, con un grosor de pared en la ecocardiografía ≥ 1,5cm al menos en un segmento miocárdico. Se consideró diagnosticados de MCH a los familiares portadores con un grosor de la pared del ventrículo izquierdo ≥ 1,3cm. A los familiares solo con pequeños cambios en el electrocardiograma (ECG) o con ECG y ecocardiografía normales, independientemente de su estado de portador, se los consideró clínicamente no afectados. A los familiares sin una evaluación diagnóstica, se los clasificó como no examinados. Se obtuvo la puntuación de riesgo de MSC (calculadora ESC) de los portadores con diagnóstico de MCH y datos clínicos disponibles. También se consideraron otros riesgos de MSC2,26: aneurisma apical del ventrículo izquierdo, MCH terminal y realce tardío de gadolinio (RTG) extenso (definido en este estudio como afección ≥ 3 segmentos cardiacos) en la resonancia magnética.
La probabilidad acumulada de incidencia de muerte cardiovascular o equivalente (MSC, descarga apropiada de un desfibrilador, muerte por insuficiencia cardiaca, muerte relacionada con accidente cerebrovascular y muerte cardiaca no especificada) y trasplante cardiaco se estimó con el método de Kaplan-Meier, y los factores se compararon mediante el método del orden logarítmico (Mantel-Cox). Se calculó la supervivencia desde el nacimiento. Se consideró que un valor de p bilateral < 0,05 indicaba significación estadística. En esta evaluación se incluyó a los portadores de p.Arg21Leu de TPM1 (probandos y familiares) y a todos los familiares de primer y segundo grado clínicamente afectados sin pruebas genéticas. La probabilidad acumulada de un diagnóstico de MCH en portadores por edad también se estimó mediante el método de Kaplan-Meier. Se calculó un logaritmo de 2 puntos de las probabilidades (puntuaciones LOD) en todas las familias utilizando el paquete PARAMLINK del software R. El modelo se estableció con θ=0, tasa de fenocopia = 0,005 y 2 valores de penetrancia diferentes: 0,80 y 0,95. Se asignó un estado indeterminado a los familiares con solo sospecha diagnóstica registrada, así como a los varones menores de 50 años y las mujeres menores de 55 años que no cumplían los criterios clínicos de MCH y más adelante podrían contraer la enfermedad.
También se recopiló información sobre la región de origen de cada probando para estimar el número de familias portadoras en diferentes regiones y la distribución geográfica de la variante en Portugal y España.
Los análisis estadísticos se realizaron con la versión 3.4.3 del software R (R Foundation for Statistical Computing, Austria) y la versión 7.7.6 de las mutaciones Health in Code (HiC) (Health in Code SA, España).
RESULTADOSSe identificó p.Arg21Leu de TPM1 en 25/10.561 (0,23%) probandos consecutivos con diferentes cardiopatías hereditarias secuenciados en nuestro centro. La variante estaba presente en 25/4.099 (0,61%) probandos de MCH y ausente en 6.462 probandos con otros fenotipos cardiacos (0/3.830 con miocardiopatías dilatada, no compactada y arritmogénica, 0/1.590 con canalopatías y 0/1.042 con otras cardiopatías hereditarias; p < 0,0001). Esta variante apareció en heterocigosis simple en 10/62.784 (0,016%) individuos de la población del programa TOPMed y en 5/120.158 individuos (0,004%) de 55-65 años de la población gnomAD (muestras no TOPMed).
Además, se incluyó a otros 6 probandos identificados en otros hospitales colaboradores. En total, se identificó a 83 portadores (el 50,6% varones) y 31 probandos. Se describieron datos clínicos detallados de 67 portadores; 44 (65,7%) se vieron afectados clínicamente (tabla 2 del material adicional y figura 1 del material adicional).
Se realizaron 28 estudios árboles genealógicos (no se comunicaron datos familiares de 3 probandos) y se documentó la cosegregación familiar de la variante con un patrón de herencia autosómico dominante (puntuación LOD combinada = 3,95). Todos los estudios genealógicos, la población de estudio, las puntuaciones LOD específicas, los datos clínicos individuales y las puntuaciones de riesgo de MSC se recogen en el material adicional.
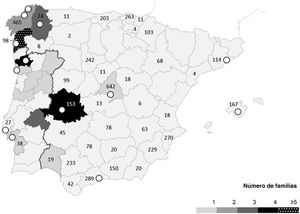
Se evaluó a probandos de 13 hospitales (4 portugueses y 9 españoles). El origen de las familias se concentró en la parte occidental de la península ibérica (la región de Braga en el norte de Portugal, y Galicia y Extremadura en España) (figura 1). No se identificó la variante p.Arg21Leu de TPM1 en muestras derivadas de hospitales de otras partes del mundo.
 de las familias portadoras de la variante p.Arg21Leu de TPM1. Mapas de Portugal y España por regiones. La concentración de familias (origen) está representada en escala de grises. Los círculos blancos indican los hospitales de referencia donde se identificó a los probandos. Los números (n) representan los estudios de miocardiopatía hipertrófica solicitados por cada región.")
Distribución geográfica (por origen) de las familias portadoras de la variante p.Arg21Leu de TPM1. Mapas de Portugal y España por regiones. La concentración de familias (origen) está representada en escala de grises. Los círculos blancos indican los hospitales de referencia donde se identificó a los probandos. Los números (n) representan los estudios de miocardiopatía hipertrófica solicitados por cada región.
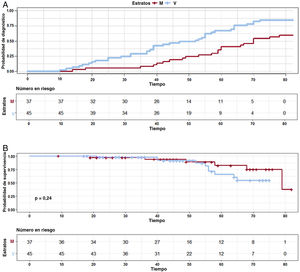
El porcentaje acumulado de diagnóstico de portadores por edad y sexo se muestra en la figura 2. A la edad de 30 años, el 25% de los varones y el 6% de las mujeres tenían diagnóstico clínico de MCH. La incidencia aumentaba a los 50 años al 50% de los varones y el 25% de las mujeres portadores. A los 70 años, aproximadamente el 17% de los varones y el 46% de las mujeres aún no estaban afectados. La media de edad en el momento del diagnóstico fue 47,3 ± 18,8 (intervalo, 11-73) años. Los individuos diagnosticados menores de 35 años fueron el 18% (12/67) de los portadores con datos detallados.
Características fenotípicas y función de supervivencia (abajo), tanto por edad como por sexo. Los varones están representados en azul y las mujeres, en rojo. M: mujer; V: varón.")
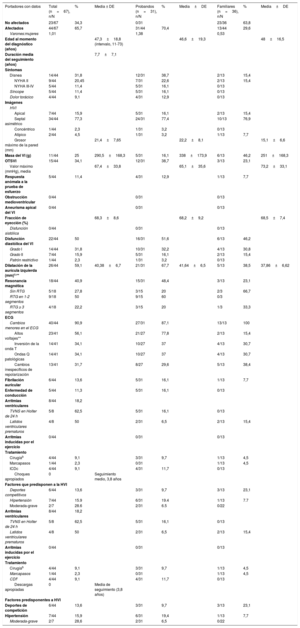
Los datos clínicos obtenidos de 67 portadores (31 probandos y 36 familiares) se resumen en la tabla 1; 44 portadores (66%; 31 probandos y 13 familiares) cumplían los criterios de diagnóstico de MCH según los criterios de hipertrofia ventricular izquierda (HVI) y 23 (34%) no se vieron afectados.
Características clínicas de p.Arg21Leu de TPM1 (n=67)
| Portadores con datos | Total (n=67), n/N | % | Media ± DE | Probandos (n=31), n/N | % | Media±DE | Familiares (n=36), n/N | % | Media±DE |
|---|---|---|---|---|---|---|---|---|---|
| No afectados | 23/67 | 34,3 | 0/31 | 23/36 | 63,8 | ||||
| Afectados | 44/67 | 65,7 | 31/44 | 70,4 | 13/44 | 29,6 | |||
| Varones:mujeres | 1,01 | 1,38 | 0,53 | ||||||
| Edad al momento del diagnóstico (años) | 47,3±18,8 (intervalo, 11-73) | 46,6±19,3 | 48±16,5 | ||||||
| Duración media del seguimiento (años) | 7,7±7,1 | ||||||||
| Síntomas | |||||||||
| Disnea | 14/44 | 31,8 | 12/31 | 38,7 | 2/13 | 15,4 | |||
| NYHA II | 9/44 | 20,45 | 7/31 | 22,6 | 2/13 | 15,4 | |||
| NYHA III-IV | 5/44 | 11,4 | 5/31 | 16,1 | 0/13 | ||||
| Síncope | 5/44 | 11,4 | 5/31 | 16,1 | 0/13 | ||||
| Dolor torácico | 4/44 | 9,1 | 4/31 | 12,9 | 0/13 | ||||
| Imágenes | |||||||||
| HVI | |||||||||
| Apical | 7/44 | 15,9 | 5/31 | 16,1 | 2/13 | 15,4 | |||
| Septal asimétrico | 34/44 | 77,3 | 24/31 | 77,4 | 10/13 | 76,9 | |||
| Concéntrico | 1/44 | 2,3 | 1/31 | 3,2 | 0/13 | ||||
| Atípico | 2/44 | 4,5 | 1/31 | 3,2 | 1/13 | 7,7 | |||
| Grosor máximo de la pared (mm) | 21,4±7,65 | 22,2±8,1 | 15,1±6,6 | ||||||
| Masa del VI (g) | 11/44 | 25 | 290,5±168,3 | 5/31 | 16,1 | 338±173,9 | 6/13 | 46,2 | 251±168,3 |
| OTSVI | 15/44 | 34,1 | 12/31 | 38,7 | 3/13 | 23,1 | |||
| Valor máximo (mmHg), media | 67,4±33,8 | 65,1±35,6 | 73,2±33,1 | ||||||
| Respuesta anómala a la prueba de esfuerzo | 5/44 | 11,4 | 4/31 | 12,9 | 1/13 | 7,7 | |||
| Obstrucción medioventricular | 0/44 | 0/31 | 0/13 | ||||||
| Aneurisma apical del VI | 0/44 | 0/31 | 0/13 | ||||||
| Fracción de eyección (%) | 68,3±8,6 | 68,2±9,2 | 68,5±7,4 | ||||||
| Disfunción sistólica | 0/44 | 0/31 | 0/13 | ||||||
| Disfunción diastólica del VI | 22/44 | 50 | 16/31 | 51,6 | 6/13 | 46,2 | |||
| Grado I | 14/44 | 31,8 | 10/31 | 32,2 | 4/13 | 30,8 | |||
| Grado II | 7/44 | 15,9 | 5/31 | 16,1 | 2/13 | 15,4 | |||
| Patrón restrictivo | 1/44 | 2,3 | 1/31 | 3,2 | 0/13 | ||||
| Dilatación de la aurícula izquierda (mm)a,** | 26/44 | 59,1 | 40,38±6,7 | 21/31 | 67,7 | 41,64±6,5 | 5/13 | 38,5 | 37,86±6,62 |
| Resonancia magnética | 18/44 | 40,9 | 15/31 | 48,4 | 3/13 | 23,1 | |||
| Sin RTG | 5/18 | 27,8 | 3/15 | 20 | 2/3 | 66,7 | |||
| RTG en 1-2 segmentos | 9/18 | 50 | 9/15 | 60 | 0/3 | ||||
| RTG ≥ 3 segmentos | 4/18 | 22,2 | 3/15 | 20 | 1/3 | 33,3 | |||
| ECG | |||||||||
| Cambios menores en el ECG | 40/44 | 90,9 | 27/31 | 87,1 | 13/13 | 100 | |||
| Altos voltajes** | 23/41 | 56,1 | 21/27 | 77,8 | 2/13 | 15,4 | |||
| Inversión de la onda T | 14/41 | 34,1 | 10/27 | 37 | 4/13 | 30,7 | |||
| Ondas Q patológicas | 14/41 | 34,1 | 10/27 | 37 | 4/13 | 30,7 | |||
| Cambios inespecíficos de repolarización | 13/41 | 31,7 | 8/27 | 29,6 | 5/13 | 38,4 | |||
| Fibrilación auricular | 6/44 | 13,6 | 5/31 | 16,1 | 1/13 | 7,7 | |||
| Enfermedad de conducción | 5/44 | 11,3 | 5/31 | 16,1 | 0/13 | ||||
| Arritmias ventriculares | 8/44 | 18,2 | |||||||
| TVNS en Holter de 24 h | 5/8 | 62,5 | 5/31 | 16,1 | 0/13 | ||||
| Latidos ventriculares prematuros | 4/8 | 50 | 2/31 | 6,5 | 2/13 | 15,4 | |||
| Arritmias inducidas por el ejercicio | 0/44 | 0/31 | 0/13 | ||||||
| Tratamiento | |||||||||
| Cirugíab | 4/44 | 9,1 | 3/31 | 9,7 | 1/13 | 4,5 | |||
| Marcapasos | 1/44 | 2,3 | 0/31 | 1/13 | 4,5 | ||||
| ICDc | 4/44 | 9,1 | 4/31 | 11,7 | 0/13 | ||||
| Choques apropiados | 0 | Seguimiento medio, 3,8 años | |||||||
| Factores que predisponen a la HVI | |||||||||
| Deportes competitivos | 6/44 | 13,6 | 3/31 | 9,7 | 3/13 | 23,1 | |||
| Hipertensión | 7/44 | 15.9 | 6/31 | 19.4 | 1/13 | 7.7 | |||
| Moderada-grave | 2/7 | 28.6 | 2/31 | 6.5 | 0/22 | ||||
| Arritmias ventriculares | 8/44 | 18,2 | |||||||
| TVNS en Holter de 24 h | 5/8 | 62,5 | 5/31 | 16,1 | 0/13 | ||||
| Latidos ventriculares prematuros | 4/8 | 50 | 2/31 | 6,5 | 2/13 | 15,4 | |||
| Arritmias inducidas por el ejercicio | 0/44 | 0/31 | 0/13 | ||||||
| Tratamiento | |||||||||
| Cirugíab | 4/44 | 9,1 | 3/31 | 9,7 | 1/13 | 4,5 | |||
| Marcapasos | 1/44 | 2,3 | 0/31 | 1/13 | 4,5 | ||||
| CDIc | 4/44 | 9,1 | 4/31 | 11,7 | 0/13 | ||||
| Descargas apropiadas | 0 | Media de seguimiento (3,8 años) | |||||||
| Factores predisponentes a HVI | |||||||||
| Deportes de competición | 6/44 | 13,6 | 3/31 | 9,7 | 3/13 | 23,1 | |||
| Hipertensión | 7/44 | 15,9 | 6/31 | 19,4 | 1/13 | 7,7 | |||
| Moderada-grave | 2/7 | 28,6 | 2/31 | 6,5 | 0/22 | ||||
CDI, cardiodesfibrilador implantado; DE, desviación estándar; ECG, electrocardiograma; HVI, hipertrofia del ventrículo izquierdo; NYHA, New York Heart Association; OTSVI, obstrucción del tracto de salida del ventrículo izquierdo; RTG, realce tardío de gadolinio; TVNS, taquicardia ventricular no sostenida; VI, ventrículo izquierdo. Prueba exacta de Fisher para el valor de p de las subpoblaciones de probandos y familiares, solo se muestra [**] cuando el valor de p <0,05 al comparar probandos y familiares.
El patrón morfológico del ventrículo izquierdo predominante fue hipertrofia septal asimétrica (34/44; 77%) y 7/44 (16%) presentaban hipertrofia apical. La figura 2 del material adicional muestra la distribución del grosor máximo de la pared del ventrículo izquierdo por sexo, genotipo y edad en el último seguimiento. Los portadores se concentraron en las HVI de 15-25 mm y las edades avanzadas. La media del grosor máximo de la pared del ventrículo izquierdo fue 21,4 ± 7,65mm. Entre los portadores diagnosticados antes de los 35 años, fue 27,04 ± 11,2mm. En el 34% de los casos (15/44) hubo obstrucción del tracto de salida del ventrículo izquierdo y en el 11% (5/44), respuesta anómala de la presión arterial a la prueba de esfuerzo. El 16% (7/44) presentaba un patrón de seudonormalización y solo 1 caso tenía un patrón restrictivo. No se observó obstrucción medioventricular del ventrículo izquierdo.
Se notificaron anomalías del ECG en el 91% de los portadores afectados; el 13,6% (6/44) de ellos presentaban fibrilación auricular y 5 portadores (11,3%; 5/44) tenían trastornos del sistema de conducción, todos casos de bloqueo auriculoventricular de primer grado. No se incluyó en este subgrupo a un portador con bloqueo auriculoventricular completo tras cirugía de la válvula mitral porque se consideró que era secundario a la cirugía; se le implantó un marcapasos. Se notificó síncope en el 11,5% (5/44) de los portadores afectados; 8 (8/44; 18%) de los portadores afectados presentaban arritmias ventriculares, 5 de ellos con taquicardia ventricular no sostenida registrada por el sistema de monitorización Holter de 24 horas. No hubo arritmias inducidas por la prueba de esfuerzo. No hubo informes de descargas apropiadas tras un seguimiento medio de 3,8 años entre los 4 pacientes varones a los que se había implantado un desfibrilador automático en prevención primaria; a 3 de ellos se los había diagnosticado en la adolescencia y recibieron los dispositivos aproximadamente 10 años después del diagnóstico. Dos portadores con desfibrilador automático tenían alto riesgo de MSC (el 9,94 y el 6,37% en 5 años) y los otros 2, puntuaciones de riesgo intermedias (ambos con el 4,59% en 5 años), sin marcadores de riesgo de MSC adicionales. Las puntuaciones de riesgo de MSC se obtuvieron en el momento de la toma de decisiones clínicas.
Cuatro portadores con MCH de inicio temprano (33%; 4/12) eran deportistas profesionales (todos jugadores de fútbol) y 2/32 (6,2%) eran portadores con inicio tardío (p=0,074) (tabla 3 del material adicional, características clínicas de los portadores de p.Arg21Leu de TPM1 diagnosticados antes de los 35 años).
Solo 2 características clínicas mostraron diferencias estadísticas significativas entre probandos y familiares (tabla 1): los probandos tenían mayores voltajes de ECG y más dilatación auricular que los familiares.
Estratificación del riesgo y eventos cardiovascularesEl análisis de supervivencia (figura 2) mostró que menos del 5% de nuestra población había sufrido muerte cardiovascular o un trasplante cardiaco a los 30 años, aunque esta cifra aumentó hasta el 10% a los 50 años en ambos sexos. El 25% de las mujeres y el 44% de los varones habían sufrido un evento cardiovascular importante a los 70 años. No se observó diferencia estadística entre sexos (p=0,24).
Se comunicaron 3 muertes cardiovasculares (1 MSC, 1 por insuficiencia cardiaca y 1 cardiaca no especificada) y 1 trasplante cardiaco (en total, 4/83; 5%) de portadores de p.Arg21Leu de TPM1. Tras una media de seguimiento de 7,7 ± 7,1 años, tampoco se notificaron MSC o trasplante de corazón. Se comunicaron otros 12 eventos cardiovasculares en familiares de primer o segundo grado sin pruebas genéticas; 5 MSC, 2 muertes por insuficiencia cardiaca, 2 muertes relacionadas con accidente cerebrovascular y 3 muertes cardiacas no especificadas. Se informó de que 5 portadores habían sufrido eventos cardiovasculares menores que no estaban incluidos en las curvas de supervivencia: 2 miectomías, 1 reemplazo de válvula mitral por obstrucción del tracto de salida del ventrículo izquierdo debido al movimiento anterior sistólico y 2 accidentes cerebrovasculares no mortales. Cada evento, la edad en el momento que ocurrió y el sexo del paciente se especifican en la tabla 4 del material adicional.
Se calculó el riesgo de MSC de todos los portadores afectados (n=44). La mayoría de ellos (77,3%; 34/44) tenían una puntuación de riesgo baja (<4% de riesgo en 5 años), el 18,2% (8/44) tenían valores intermedios (el 4-6% de riesgo en 5 años) y solo el 4,5% (2/44) tenía un riesgo alto (> 6% de riesgo en 5 años). La figura 3 del material adicional muestra que los portadores se concentraron en las franjas de bajo riesgo de MSC y más edad. A 18 portadores afectados (18/44; 41%) se les había realizado una resonancia magnética; 5/18 (27,8%) no mostraron RTG, 9/18 (50%) presentaron RTG en 1 o 2 segmentos cardiacos y 4/17 (22,2%), RTG en al menos 3 segmentos. No se observó aneurisma apical del ventrículo izquierdo ni MCH en fase terminal.
Portadores homocigotos y otras variantes genéticasPresentaban genotipos complejos 15 individuos (15/67; 17,9%) de 10 familias; 4 tenían p.Arg21Leu de TPM1 en homocigosis (4/67; 6%) y 12, una variante genética adicional (12/67; 17,9%); uno de los portadores homocigotos también tenía la variante patogénica p.Asp75Asn de MYBPC3. Las características clínicas de los portadores homocigotos se describen en la tabla 5 del material adicional.
Se identificaron 5 variantes genéticas en el gen MYH7 (p.Gly741Arg y p.Thr1019Asn, clasificadas respectivamente como variantes patogénicas/probablemente patogénicas; p.Lys351Asn, p.Tyr582Cys y p.Leu1333Val como variantes de significado clínico incierto). Se identificaron otras variantes sarcoméricas patogénicas: p.Met281Thr de TPM1, p.Arg278Cys de TNNT2 y p.Met173Val de MYL3. Cada una de estas variantes se identificó en diferentes familias. Todas estas variantes se han notificado con muy baja frecuencia alélica (<0,01%) en la población general, excepto p.Lys351Asn de MYH7. Consulte la descripción complementaria de cada variante en la tabla 6 del material adicional, que muestra las características clínicas de los portadores con otra variante genética.
No se comunicó muerte cardiovascular ni trasplante de corazón en los portadores con otra variante genética confirmada. Se notificaron 2 MSC de parientes de primer grado sin pruebas genéticas en familias con otra variante genética patogénica (p.Gly741Arg de MYH7 y p.Met281Val de TPM1 respectivamente).
DISCUSIÓNEn este artículo se presenta la mayor población con MCH que porta la misma variante de TPM1 que se haya descrito hasta la fecha. Este estudio muestra que la variante p.Arg21Leu de TPM1 está considerablemente sobrerrepresentada en nuestra cohorte de MCH en comparación con las poblaciones de control, y la cosegregación familiar de la variante con la enfermedad se documentó con una puntuación LOD significativa27. Teniendo en cuenta estos resultados, hay criterios suficientes para clasificar ahora la p.Arg21Leu como claramente patogénica (tabla 7 del material adicional).
Se identificó la p.Arg21Leu de TPM1 exclusivamente en pacientes con el fenotipo de MCH. La mayoría de los individuos de nuestra población eran portadores heterocigotos simples con MCH de inicio tardío, fenotipo de leve a moderado, evolución clínica asintomática y un número bajo de muertes cardiovasculares y trasplantes de corazón. La incidencia anual de eventos cardiovasculares fue del 0,25% en el análisis de supervivencia, lo que indica un pronóstico para p.Arg21Leu de TPM1 mejor de lo esperado para la propia enfermedad (≅0,5%/año)28,29. Las puntuaciones de riesgo de MSC estuvieron predominantemente en la banda de riesgo bajo, seguidas aproximadamente del 20% de los casos con puntuaciones intermedias. Solo 2 portadores tenían un riesgo de MSC alto.
En comparación, la variante fundadora finlandesa de TPM1, p.Asp175Asn, se describió en la bibliografía como asociada con un fenotipo de MCH leve-moderada y un pronóstico favorable; no obstante, se notificó mayor penetrancia en la edad adulta (91-95%), sobre la base de 2 cohortes más pequeñas no relacionadas13,14. Otras variantes en los genes MYBPC3 y MYH7 notificadas anteriormente como efectos fundadores en poblaciones de MCH han mostrado peor pronóstico que la p.Arg21Leu de TPM117–21. La media de edad en el momento del diagnóstico de los individuos portadores de algunas de estas variantes también fue menor, aproximadamente una década menos.
Los portadores de p.Arg21Leu en TPM1 con MCH de inicio temprano eran predominantemente varones. En este grupo, no se informó ninguna otra variante genética patogénica (excepto 1 que tenía una variante de MYH7 de importancia clínica desconocida). Sus puntuaciones de riesgo de MSC eran superiores a la media general y tenían una HVI más prominente (ambas miectomías descritas en nuestro estudio se encontraron en individuos de este grupo), pero no se documentó muerte cardiovascular ni trasplante de corazón entre los portadores diagnosticados antes de los 35 años. Otros grupos han descrito una evidente heterogeneidad clínica intrafamiliar, similar que con otras variantes de MCH en TPM14–11, con MSC de individuos con MCH de inicio temprano. En cambio, no hubo eventos CV importantes entre nuestros portadores con MCH de inicio temprano; sin embargo, hubo 1 MSC de un familiar de primer grado de 19 años no genotipificado (debe tenerse en cuenta que se identificó otra variante patogénica de MYH7 en esta familia) y se comunicó otro caso de MSC de 1 pariente de segundo grado de 21 años no genotipificado. No se informó de MCH en ninguno de estos portadores.
En este estudio se notifica un número considerable de portadores no afectados de edades avanzadas en nuestras familias y también en la población general, lo que indica una penetrancia incompleta. Este inicio tardío de la enfermedad y la penetrancia incompleta de la variante podrían suponer un reto a la hora de demostrar la cosegregación familiar de una variante rara, como p.Arg21Leu de TPM1, lo que requirió la agrupación de un mayor número de familias, como se llevó a cabo en esta investigación.
Portadores homocigotos y variantes genéticas adicionalesCasi el 18% de los portadores presentaban una segunda variante genética, porcentaje superior al descrito en la bibliografía (5%) sobre MCH2. Estudios previos han demostrado que el número de variantes genéticas puede ser un factor determinante de la gravedad de la enfermedad30,31. Sin embargo, nuestros datos demuestran que la existencia de una segunda variante no se asocia necesariamente con un fenotipo más grave. La mayoría de los portadores con una variante genética adicional presentaban fenotipos leves o no estaban afectados, lo que podría explicarse por la existencia de 2 variantes con penetrancia tardía o incompleta. La interpretación clínica de los hallazgos genéticos requiere considerar las características clínicas y la penetrancia descritas específicamente de cada una de las variantes identificadas.
Además, también se podría plantear la hipótesis de que la coexistencia de otra variante genética sería necesaria para la penetrancia (manifestación de la enfermedad) de otra variante rara identificada en la población general, pero no ha sido posible comprobar esta asociación estadísticamente en nuestra población.
Varios estudios han asociado variantes sarcoméricas en homocigosis con presentaciones más graves en MCH que cuando estas variantes se identifican en heterocigosis simple32,33. Los portadores homocigotos de p.Arg21Leu de TPM1 en nuestro estudio mostraron, en general, un fenotipo más pronunciado que los portadores heterocigotos simples (especialmente si se tiene en cuenta la existencia de disfunción diastólica moderada-grave y arritmias ventriculares); sin embargo, estos portadores homocigotos mostraron manifestaciones de la enfermedad de inicio tardío, lo que respalda la opinión de que esta variante probablemente no esté asociada con un fenotipo para el curso de vida.
p.Arg21Leu de TPM1 como variante fundadoraLa distribución geográfica de las familias portadoras mostró una concentración predominante en la parte occidental de la península ibérica, especialmente en las regiones españolas de Galicia y Extremadura, y el norte de Portugal (figura 1). Estos territorios comparten factores históricos y geopolíticos comunes que se remontan al siglo xi o incluso antes34. Se identificó a 4 probandos en hospitales colaboradores ubicados fuera de estas zonas, pero se comunicó que el origen de las familias se encontraba en la región fundadora. Además, la identificación de portadores homocigotos con progenitores no consanguíneos también reflejaría la mayor prevalencia de la variante en regiones específicas.
Un estudio reciente ha descrito una variante fundadora del gen GLA en la misma región del norte de Portugal, donde también se ha identificado gran número de familias portadoras de la variante p.Arg21Leu de TPM135. Los autores demostraron que ello estaba relacionado con las particularidades culturales y socioeconómicas (es decir, un alto nivel de endogamia a causa de matrimonios dentro del mismo estrato social) existentes entre los siglos xvii y xix en esta región. Consideraron que los factores sociales y la penetrancia tardía de la variante perpetuaron la transmisión de la enfermedad en esa región hasta la época contemporánea. Se ha conjeturado que estas características podrían explicar la distribución observada de la variante p.Arg21Leu de TPM1.
La existencia de nuestra variante exclusivamente en latinos de poblaciones gnomAD y TOPMed podría reflejar un posible ancestro común de España y Portugal, dado que estos países tuvieron un papel fundamental en la colonización de Latinoamérica. En comparación, se comunica que otra variante de TPM1, p.Asp175Asn, descrita como efecto fundador en Finlandia, se encuentra predominantemente en individuos finlandeses europeos en la base de datos gnomAD.
LimitacionesEste estudio de correlación entre genotipo y fenotipo tiene algunas limitaciones potenciales. En primer lugar, no se comunicó ninguna evaluación clínica ni genética en un gran número (n=106) de familiares de primer grado identificados en los estudios familiares. Esto puede estar relacionado con la baja repercusión clínica de la variante y la percepción de riesgo de los médicos y las familias portadoras. Algunas pruebas se realizaron solo en un pequeño número de casos, como la resonancia magnética cardiaca, lo que limita nuestra capacidad para evaluar otros marcadores de riesgo. Además, la curva de Kaplan-Meier presentada en este estudio que muestra la edad en el momento del diagnóstico no debe tomarse como la edad real al inicio de la expresión de la enfermedad, sino solo como el momento en que se estableció el diagnóstico.
Aunque se describe una población relativamente grande que porta la misma variante, las cifras aún son demasiado pequeñas para permitir conclusiones definitivas. En este sentido, un estudio comparativo de las características clínicas de p.Arg21Leu de TPM1 frente a otras variantes genéticas de este mismo gen puede ofrecer más evidencia para lograr una estratificación del riesgo más asertiva en la MCH. Finalmente, es necesario realizar más estudios para caracterizar mejor el efecto fundador al que este artículo apunta, como los estudios de haplotipos. Sin embargo, creemos que los datos epidemiológicos disponibles son suficientes para respaldar un efecto fundador.
CONCLUSIONESLa disponibilidad de un elevado número de portadores con la misma variante de TPM1 de un área geográfica definida en Galicia, Extremadura y norte de Portugal permitió la correlación genotipo-fenotipo que reveló que la variante p.Arg21Leu es patogénica y se asocia con MCH de inicio tardío, penetrancia incompleta y una evolución clínica generalmente favorable.
- –
TPM1 es uno de los principales genes de la MCH y justifica el 1-5% de los casos de la enfermedad.
- –
La información clínica disponible sobre los portadores de TPM1 es bastante escasa en la bibliografía. Por lo tanto, los estudios de correlación genotipo-fenotipo de poblaciones con MCH portadoras de la misma variante sarcomérica podrían ser la oportunidad ideal para comprender mejor el perfil clínico y el pronóstico asociado.
- –
Esta es la mayor población de MCH descrita con la misma variante de TPM1, lo que posiblemente constituya un efecto fundador en Portugal y España.
- –
p.Arg21Leu de TPM1 es una variante patogénica de MCH con inicio tardío/penetrancia incompleta y un pronóstico generalmente favorable.
- –
Este estudio podría ser útil para la interpretación clínica de hallazgos genéticos en MCH y para la elaboración de políticas sanitarias en estas regiones.
Este artículo no ha contado con financiación.
CONFLICTO DE INTERESESA. Lamounier Junior y M. Ortiz han recibido honorarios personales de Health in Code SL, al margen de este estudio. L. Monserrat Iglesias es accionista de Health in Code SL.