Keywords
INTRODUCTION
From 1961, when right ventricular arrhythmogenic dysplasia was first described by Dalla Volta et al1 and subsequently characterized by Fontaine et al2 in 1977, until present, when the condition has been included in the World Health Organization classification of cardiomyopathies,3 many reports of the disease have appeared in the literature.4,5 Initially, reports focussed on the arrhythmic substrate of certain regions of the right ventricle (RV), the so-called "triangle of dysplasia," though lines of investigation have since broadened to include diffuse manifestations in the RV, involvement of the left ventricle only and biventricular involvement in dilated phase, which are often indistinguishable from dilated cardiomyopathy (DMC).6
GENETICS AND PATHOGENESIS
A familial history of the disease is present in up to 50% of the patients. Inheritance is of the dominant autosomal type with variable expression and incomplete penetrance (30%).7 The recessive autosomal form has also been described in association with Naxos disease8,9 and mutations of the gene encoding desmoplakin (DSP).10
Nine loci associated with arrhythmogenic right ventricular cardiomyopathy (ARVC) have been described. Currently, 3 gene mutations causally related to the disease are known. Table 1 summarizes the loci isolated by chromosome mapping techniques.

Although the cause of ARVC is still not known, several theories have been suggested. In the inflammatory theory--supported by the appearance of inflammatory infiltrates in autopsy cases--, myocardial damage is explained by a continuous cycle of damage and repair that simulates chronic myocarditis. In the differential diagnosis of ARVC, chronic myocarditis should be included with RV involvement only, so complicating the investigation of the cause of the disease. According to the genetic theory, mutations of genes that encode specific proteins would give rise to myocardial "dystrophy." Furthermore, recent discoveries of disease-causing mutations have led to new pathogenetic theories based on intercellular mechanical stress. Descriptive studies indicate that progressive myocardial replacement by fatty and fibrous tissue cells occurs after exaggerated and inappropriate apoptosis.11-13 Animal models lend support to the imbalance caused by cellular mechanical stress as a trigger of apoptosis.14,15
Cell-Cell Adhesion Abnormalities
A possible mechanism for ARVC is progressive loss of myocytes secondary to structural alterations--a mechanism that has also been described in the genesis of DMC.16
Cell-cell junctions are formed by proteins. These include plakoglobin (JUP) and DSP which play a key role in the transduction of mechanical stress and cell-cell communication.17 Both proteins are present in cardiac myocytes and in epidermal adhesion.18,19 Plakoglobin is a cytoplasmic protein that forms part of both desmosomes20 and adherens junctions. It helps link intermediate and cytoskeletal actin filaments with the transmembrane complexes that connect adjacent cells. The mutated form of JUP21 favors an unstable cell substrate.9 Desmoplakin also regulates expression of the antiapoptotic protein Bcl-2.22
Desmoplakin is a component of the desmosome plaque where its functions include anchoring the intermediate filaments to the plasma membrane. The protein is also essential for maintenance of cell integrity and it acts as an anchor for desmin in Purkinje cells.23
The first mutation of the gene that encodes JUP, reported in the long arm of chromosome 17,8,24 is related to transmission of the recessive form of Naxos disease (nonkeratolytic palmoplantar keratoderma, woolly hair, and ARVC). Now other mutations are beginning to be found. An S299R mutation in exon 7 of DSP has been described in a family with ARVC with dominant autosomal inheritance.25 The association of skin disorders, woolly hair, and heart disorders has been described in recessive forms of both ARVC (Naxos disease) and DMC.26
These proteins therefore have a structural function similar to others described in the genesis of DMC.27
Excitation-Contraction Coupling Disorders
The cardiac ryanodine receptor (RYR2) forms part of the structure that regulates the calcium channels of the sarcoplasmatic reticulum. These channels have to be functioning correctly for excitation-contraction coupling and calcium homeostasis in monocytes. Six mutations of RYR2 have been described in different families: 3 encode the terminal amino acid and the other 3 the protein binding site.28,29 Greater susceptibility to exercise-induced ventricular tachycardias has been reported in these families.
In 1995 Rampazzo30 described a second locus associated with what he identified as a new variant of ARVC, ARVC2, which is characterized by catecholaminergic-induced ventricular tachycardias. Mutations have been detected in this gene in catecholaminergic polymorphic ventricular tachycardia, familiar polymorphic ventricular tachycardia, and ARVC2.29,31,32 These, unlike ARVC2, do not show replacement with fibrofatty tissue. RYR2 mutations could increase the cytosolic concentration of calcium and trigger programmed cell death.15,33-38 Decoupling of excitation-contraction would also occur, provoking arrhythmias.
Further reports of ARVC mutations advance our understanding of the wide variety of presentations of the disease and its variability from one family to another and within the same family, but they also generate controversy because some entities should perhaps not be considered as ARVC. In particular, exercise-induced tachycardias are probably independent of ARVC.
An important step in the understanding and characterization of ARVC will certainly be the pooling of resources to set up a gene bank.11,20
CLINICAL PRESENTATION AND NATURAL HISTORY
Arrhythmogenic right ventricular cardiomyopathy usually becomes manifest in adolescents and adults and affects more men than women. Prevalence varies widely according to the study populations, and the geographic distribution of the disease is controversial. In Italy, the prevalence in Veneto region is estimated to be 1 case per 1000 or 10 000 people. Corrado et al39,40 reported that ARVC could be the cause of up to 20% of sudden deaths (SD) in young adults, and that it is the most common cause of SD in Italian adults. In the United States, ARVC represents 5% of SD in those under 65 years of age41 and it may be the cause of 3%-4% of SD in athletes.42 It could also be a frequent cause of perioperative deaths in subjects with no evidence of prior structural heart disease.43-45 In Spain, ARVC is reported to be a common cause of SD in young men.46
Clinical manifestations vary and depend on cardiac instability and progressive venous dysfunction. Arrhythmogenic right ventricular cardiomyopathy may present without symptoms, or it may manifest itself first as SD, ventricular and supraventricular arrhythmias, or right or biventricular heart failure.
In ARVC, an imbalance in adrenergic innervation has been described as a possible factor in the genesis of arrhythmias. Thus, the propensity to ventricular arrhythmias would increase on exposure to catecholamines, particularly during exercise.47
Although the information relating the natural history of the disease is limited, four stages are generally identified:
1. The early or silent phase, generally asymptomatic, although it may first manifest itself as SD.48
2. The unstable phase with predominance of symptomatic arrhythmias, generally of complete left branch bundle block type, highly suggestive of right ventricular origin.
3. Phase of right ventricular failure with relative conservation of left ventricular (LV) function.
4. Final phase with progressive biventricular dilatation, often indistinguishable from DMC. The most common complications of this stage are thromboembolism and atrial fibrillation.49
Left Ventricular Involvement
Left ventricular involvement is usually associated with severe disease progression. Populations with long-term follow up have clarified and defined LV involvement in ARVC. In a study of 42 patients, 76% showed left ventricular involvement. The involvement varied according to age and was associated with arrhythmic events, more severe cardiomegalia, inflammatory infiltrates and heart failure.50
Cases of arrhythmogenic LV cardiomyopathy with involvement of the left ventricle only have also been reported. The histologic LV characteristics mimicked those usually seen in the RV.51,52
DIAGNOSIS
The definitive diagnosis of ARVC requires histologic confirmation from surgical or autopsy samples showing the presence of transmural replacement with fibrofatty tissue. The patchy and progressive nature of the disease limits the usefulness of endomyocardial biopsy.
No single test can confirm diagnosis of ARVC.3,7 Diagnosis is established after functional, morphological, and electrocardiographic evaluation to investigate major and minor criteria by which the disease is recognized (Table 2).

Diagnosis is confirmed if 2 major criteria, 1 major criteria and 2 minor ones, or 4 minor ones are met. The usefulness of these criteria has been proven prospectively.
Table 3 presents the criteria proposed by Hamid et al53 to increase the diagnostic sensitivity of first-degree family members.

Currently, several longitudinal and prospective international registries are in progress in Europe49,50 and North America.54 These registries aim to provide an analysis of the validity of the current diagnostic criteria and the incorporation of contributions from phenotype-genotype associations to improve diagnostic efficiency.
DIAGNOSTIC TESTS
Electrocardiogram
The progressive nature of the disease has been demonstrated in prospective studies, thus the initial electrocardiogram (ECG) may be normal. In a series of 74 patients with VT and ARVC, 40% had a normal ECG in the first year after the VT episode, but after 5 years of follow up, only 8% had normal ECG, and after 6 years, no patients had normal ECG.55
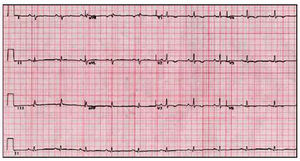
The most common ECG abnormality is T-wave inversion (V1-V3)--present in up to 50% of the subjects.56 Similar abnormalities in other leads indicates additional LV impairment57 (Figure 1).

Fig. 1. Electrocardiogram of a patient with arrhythmogenic right ventricular cardiomyopathy and left ventricular involvement.
Various depolarization abnormalities have been described in ARVC such as partial right bundle branch block (18%)--more common than complete right bundle branch block (15%)--; QRS interval duration of more than 110 ms in V1 and V2--a more specific finding--58; and the appearance of epsilon waves (Figure 2), which are seen in 30% of the patients.59 Epsilon waves may go unnoticed, though the sensitivity for their detection can be increased by suitably preparing the skin and recording the ECG at double speed and double amplitude.60 The waves appear at the end of the QRS complex and at the start of the ST segment and correspond to delayed electric potentials of small amplitude that originate in areas of healthy tissue surrounded by fibrofatty infiltrate.61

Fig. 2. Epsilon wave in lead V1.
The presence of late potentials in the signal-averaged ECG (SAECG) is equivalent to epsilon waves, and is related to myocardial fibrosis. Between 50% and 80% of patients with VT present SAECG abnormalities,62,63 although this test may appear normal when disease is very localized.
Echocardiography
The imaging technique of choice is echocardiography because it is noninvasive and allows disease progression assessment and to study family members. Differential diagnosis is also possible with respect to other entities associated with arrhythmias and RV dilatation such as atrial shunts, tricuspid or pulmonary valve disease, RV infarction, congenital absence of the right pericardium, the Ebstein anomaly and partial anomalous pulmonary return.64,65
The echocardiographic findings described include RV dilatation and hypokinesia, end-diastolic aneurysms and dyskinesia of the inferobasal segment. Function should be evaluated in different regions of the RV given the segmental nature of the disease. The ratio between the right and left ventricular volumes has been found to be useful in diagnosis. Other findings are increased echogenicity of the moderator band, prominent apical trabeculae,66 and tricuspid valve prolapse.
With the advance of imaging techniques and the development of software for objective analysis of the images obtained, echocardiography can help detect early abnormalities as well as progression of the disease. Tissue Doppler spectroscopy is also considered useful for detection of abnormalities in diastolic tricuspid annular motion.67
The benefit of echocardiographic contrast for diagnosis of ARVC has yet to be demonstrated, although preliminary results are promising.
Right Ventriculography
This technique is considered the gold standard for evaluation of RV function.59,66 In studies reported in the literature, specificity is as high as 90% for detection areas of akinesia in the so-called triangle of dysplasia. Nevertheless, the method is not widely used because of interobserver variations and because it is an invasive technique.
Cardiac Magnetic Resonance
Cardiac magnetic resonance is a highly sensitive noninvasive technique that can probe both functional and structural abnormalities. Unlike echocardiography, is not limited by the acoustic window and it can also be used for tissue characterization. The technique ideally offers decisive advantages: it can characterize fibrosis--potentially useful for diagnosis of the initial stages--and fatty tissue infiltration, and detect structural anomalies (Figure 3). Identification of fatty tissue in the RV myocardium does not in itself imply diagnosis of ARVC,6 as healthy individuals can also show fatty infiltration, mainly at the anterior wall of the RV. Despite its advantages it does have some limitations due to interobserver variations,61,68,69 and decreases in the thickness of the RV wall, which may be such that the spatial resolution is no longer high enough to quantify ventricular thickness (artefacts due to movement). Difficulties also arise in the evaluation of patients with arrhythmias and even those with numerous extrasystoles.

Fig. 3. Cardiac magnetic resonance image of bumps in the free wall of the right ventricular. RV indicates right ventricle; LV, left ventricle.
Therefore, cardiac magnetic resonance is a technique in the process of evaluation. For the time being, the results from such examinations should be considered in conjunction with other diagnostic tests.
Nuclear Medicine
Apart from the obvious contributions for evaluation of ventricular volume and function, scintigraphy with 131I-meta-iodobenzylguanidine at a molecular level has allowed identification of sympathetic innervation abnormalities.70 Likewise, other studies have shown that there are fewer beta-adrenergic postsynaptic receptors in the myocardium.47
Endomyocardial Biopsy
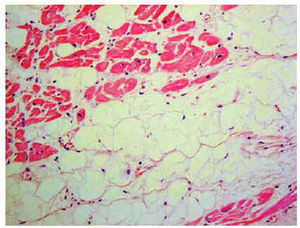
Histological diagnosis is definitive71 (Figure 4). However, the usefulness of endomyocardial biopsy is shrouded in controversy because of the segmental nature of the disease, limited septal involvement (samples are usually obtained from the septum), a high rate of complications (tamponade and perforation) related to narrowing of the myocardial wall and the greater technical difficulty (often samples are obtained from uncommon sites).7

Fig. 4. Histologic sample: fibrofatty infiltration of the right ventricle.
Signal Averaged Electrocardiogram, Holter Monitoring, and Exercise Testing
Slow conduction zones generate late postpotentials. Studies suggest that the prevalence of these potentials varies with a sensitivity of 45% and a high specificity (95%).72 QT dispersion greater than 40 ms, a measure of lack of homogeneity of ventricular repolarization, has been proposed in combination with SAECG to identify patients with ARVC with VT of right-side origin.
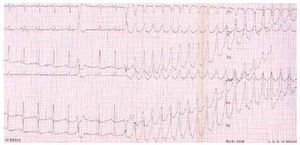
The exercise test and Holter monitoring show arrhythmias which prove diagnostic but the prognostic value of a normal outcome is very limited (Figure 5).

Fig. 5. Ventricular tachycardia with left bundle branch block morphology induced during exercise test.
INTERNATIONAL REGISTRIES
Arrhythmogenic right ventricular cardiomyopathy is currently subject to intense investigation. There are 2 main registries currently in use, one in North America (www.arvd.org) and the other in Europe (www.arvd.net). The objectives and study protocols aim to extend our knowledge on ARVC.
TREATMENT, RISK STRATIFICATION, AND PROGNOSIS
Arrhythmogenic right ventricular cardiomyopathy is a common cause of SD. The appearance of SD is not related to disease progression, which means that SD may be the first manifestation of the disease. Although prospective studies of survival are not yet available, the annual mortality rate without treatment is 2.5%-3% and 1% for individuals receiving pharmacological treatment.73,74
Treatment should be individualized. Different therapeutic options include pharmacological treatment, an implantable cardioverter defibrillator (ICD), radiofrequency ablation, and transplantation.
No pharmacological treatment, either alone or in association with other drugs, has been shown to offer full protection against SD. For symptomatic treatment of arrhythmias, essentially triggered after catecholaminergic stimulus, beta-blockers have been shown to be particularly effective.75,41
The use of drugs such as sotalol and amiodarone, alone or associated with beta-blockers, has proved effective in the treatment of symptomatic arrhythmias.61,76 In particular, a study showed that sotalol at high doses was able to suppress ventricular arrhythmias in up to 84% of patients who had presented inducible ventricular arrhythmias during the electrophysiological study.77
Implantation of an ICD should be considered in high risk patients.
In a recent prospective study, 132 patients with ARVC received an ICD for the following indications78: prior history of cardiac arrest, VT with hemodynamic intolerance (syncope or circulatory collapse), VT without hemodynamic intolerance, unsustained VT detected during Holter monitoring or an exercise test, or family history of more than one SD in the family. After a mean follow up of 3.3 months, 4 deaths had been reported, 1 due to arrhythmia after refractory ventricular fibrillation (VF), 2 related to infection of the ICD pouch, and another after progressive heart failure. Moreover, during follow-up, 3 transplants were needed, 2 because of biventricular failure and 1 due to untreatable VF. Of the 132 patients, 64 (48%) received a total of 1271 appropriate discharges. The ICD discharge rate in the study group was of 15% a year. The time from implantation until the first ICD discharge varied greatly (from 2 months to 8 years). There were no significant differences with regard to use of drugs between the group with appropriate ICD discharges and the 68 patients (52%) who received no ICD discharges.
According to statistical analysis, previous cardiac arrest or VT with compromised hemodynamics, young age, and decreased LV ejection fraction were possible fibrillation/ventricular flutter risk markers. Unexplained syncope was almost statistically significant. Patients who received an ICD because of a familial history of SD or VT without compromised hemodynamics did not show significant beneficial results. The lack of significance in the case of familial history could be due to the small number of patients who received an ICD for this reason alone.
Retrospective studies have shown different factors related to SD such as young age, history of unexplained syncope, family history of SD, participation in competitive sports, diffuse right ventricular dilatation and involvement of the LV79-81 (Table 4).

With regard to the technical difficulties, it is important to emphasize that the site of electrode implantation is crucial for appropriate detection of ventricular arrhythmias.82
Other invasive therapeutic options include radiofrequency ablation and surgical treatment of arrhythmias.
Radiofrequency ablation is used in refractory cases although its use is limited because of the thinness of the RV and the progressive nature of ARVC. With regard to the prognostic value, even if VT cannot be induced with or without pharmacological therapy, the risk of SD is not reduced.83
Surgical treatment of arrhythmias in refractory cases needs a ventriculotomy or isolation of the RV to isolate the macroreentrant circuit.2 The risk of complications, the high rate of recurrence of ventricular arrhythmias and subsequent hemodynamic abnormalities are increasingly limiting its use, although transplantation is an option to consider in late phases of the disease.
Exercise and Sudden Death
Those who practice competitive sports are at greater risk of SD.84 However, the incidence of ARVC in autopsy series varies greatly and depends on the site and circumstances of ARVC. Recent results85 from a retrospective study of 200 autopsies of individuals with ARVC who died suddenly suggest that the SD occurs more often during sedentary activities. However, according to repeated observations by different groups, ARVC is a common cause of SD, particularly in male athletes (≤35 years).39,42,46 The group in Padua78 published one of the few prospective studies to investigate the appearance of SD in young athletes of both sexes in comparison with a similar population who did not participate in competitive sports. The findings indicate that sport itself is not the cause of higher mortality, but rather acts as a trigger for cardiac arrest in entities such as ARVC, premature cardiovascular disease and congenital coronary abnormalities.
CONCLUSION
After recent discoveries, arrhythmogenic right ventricular cardiomyopathy seems more complex. Although its incidence was recently estimated to be 1:5000, the epidemiological impact of this disease is enhanced by its familial nature and hereditary pattern which requires a multidisciplinary approach and close monitoring of family members. International efforts to extend the information available at the clinical, diagnostic and genetic level require cooperation of many countries. Finally, although pharmacological treatment has been shown to be effective in most patients, use of an ICD should be considered, particularly among those with a high risk profile.
Funding: José M. García-Pinilla received a grant from the Spanish Society of Cardiology for short stays abroad in 2002. María T. Tomé Esteban received a grant from the Spanish Society of Cardiology for postgraduate training abroad in 2002 and is currently a Clinical Research Fellow funded by the British Heart Foundation.
Correspondence: Prof. W.J. McKenna.
Cardiology in The Young. The Heart Hospital.
16-18 Westmoreland Street. London W1G 8PH. United Kingdom.
E-mail: william.mckenna@uclh.org