Keywords
INTRODUCTION
The right ventricle and pulmonary circulationhave traditionally been considered to play very mucha secondary role in cardiology in general and heartfailure in particular.1
However, currently, we can affirm that both rightventricular (RV) function and pulmonary circulationstatus are of utmost importance in cardiology.Specifically, in patients with heart failure, pulmonaryhypertension (PH) and RV function are determiningfactors in the clinical picture, and essential prognostic variables and indispensable for some of the mostimportant therapeutic decisions.
It is estimated that approximately 60% of patientswith severe left ventricular systolic dysfunction and70% of those with isolated diastolic dysfunctionexperience PH.2 Given the high prevalence of theseconditions, we can affirm that PH secondary to leftheart disease, and specifically to heart failure, is oneof the most common types of PH.
DEFINITIONS
The new clinical practice guidelines for thediagnosis and treatment of pulmonary hypertensiondefine PH secondary to left heart disease as apathophysiological and hemodynamic entity thatmay present as any one of a wide variety of clinicalentities that affect the left heart chambers andstructures.3
PH secondary to left heart disease represents group2 of the new modified Dana Point classification(Table 1).4 It is one of the main representatives ofthe so-called non-PAH.5

The definition of PH secondary to left heartdisease is coupled with the need for a hemodynamicstudy to confirm that the mean pulmonary arterypressure (mPAP) is ≥25 mm Hg at rest and that thepulmonary wedge pressure (PWP) is >15 mm Hg.
The 25 mm Hg cut-off has probably been chosento make the hemodynamic limit more uniform forall forms of PH and because it is the value used inclinical trials and PH registries. However, the recentreassessment of the hemodynamic data availablefor healthy subjects has shown that normal mPAPat rest is 14 (3) mm Hg, with a maximum limitof normal rarely in excess of 20 mm Hg.6 In fact,previously accepted definitions set the upper limitof normal for mPAP at 19 mm Hg.7 Therefore,the significance of mPAP lying between 20 and 24mm Hg is not clear and patients with a mPAP in thisrange need to be assessed further in epidemiologicalstudies.3
PATHOPHYSIOLOGY OF PULMONARYHYPERTENSION IN HEART FAILURE
The lung, in addition to oxygenating venousblood, has the unique characteristic of being theonly organ that spends the entire cardiac cycle at "low pressure," even when exercise causes up to a5-fold increase in cardiac output. This is possiblethanks to the enormous reserve of the pulmonaryvascular bed. This capacity also contributes to theregulation of left ventricular filling by maintainingthe transpulmonary gradient (TPG) (TPG=mPAPPWP) within normal values, close to 5-7 mm Hg.
Passive and Reactive PulmonaryHypertension
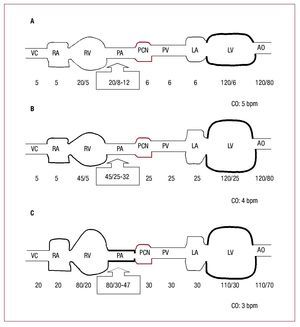
PH in patients with heart failure may have passiveor reactive PH (and in the latter case, reversible orirreversible), although in clinical practice it is usuallymixed, that is, passive with a reactive component(Figure 1).

Figure 1. Schematic diagram ofpulmonary circulation. A: healthyindividual. B: heart failure and mild "passive" pulmonary hypertension. C:advanced heart failure and severe"reactive" pulmonary hypertension. Thepressures (mm Hg) are presented from thevena cava (VC), right atrium (RA), rightventricle (RV), pulmonary artery (PA),pulmonary capillary bed (PCB), pulmonaryvein (PV), left atrium (LA), left ventricle (LV), and the aorta (AO). CO indicatescardiac output in each case.
When elevated PWP occurs in patients withsystolic, diastolic, or mixed heart failure (Table 2),there is initially a "passive" increase in mPAP inorder to maintain normal TPG sufficient to facilitateflow from pulmonary circulation to the left side ofthe heart.

However, chronically elevated PWP isaccompanied by a "reactive" increase in mPAP,in addition to the passive component, and soTPG increases. Clinical practice guidelines for PHintroduce an additional hemodynamic definition tothis entity3: Passive PH when TPG is ≤12 mm Hgand reactive or disproportionate PH when TPG is >12 mm Hg.
The reactive component, in turn, has a dynamic orfunctional component resulting from vasoconstrictivestimuli and a fixed component. The first is generally reversible with vasodilator administration.However, the fixed component reflects remodelingof the pulmonary artery muscle, essentially in theform of medial hypertrophy and, to a lesser extent,intimal fibrosis. Thus, the pulmonary artery vesselpartially loses its vasodilatory capacity. Dependingon the extent of this loss, reactive PH will be moreor less reversible (or fixed) when vasodilator drugsare administered.
Reactive PH eventually leads to RV dysfunction(as pulmonary pressure is the main determinant ofafterload) and, finally, decreased cardiac output andright heart failure.
Right Ventricle
The RV, in the context of heart failure, may beaffected principally by the primary heart disease(idiopathic or ischemic cardiomyopathy) and/or byincreased afterload leading to PH and its progression.As the RV becomes dilated in response to poorlytolerated pressure overload, restrictions imposedby the pericardium and the muscle fibers commonto both ventricles (interventricular interdependencerelationship) limit further RV dilation. This leadsto a steeper gradient of the RV diastolic pressure-volume curve, such that a larger increase in RVpressure does not correspond to greater stretchingof its free wall. At the same time, displacement ofthe ventricular septum reduces the left ventricularejection fraction. Together, these 2 events lead toa net decrease in cardiac output. Once the cardiacoutput begins to decrease, ventricular failureprogresses rapidly. After the decrease in cardiacload comes systemic hypotension; this reduces theRV perfusion pressure and favors ischemia of thefree wall. RV ischemia further reduces contractilefunction, with still larger decreases in cardiac load,and a rapid downward spiral is initiated resulting inhemodynamic collapse.8
Protection Against Pulmonary Edema
The development of fixed PH is one of theprotective mechanisms against pulmonary edemain presence of chronically elevated left ventricularpreload. Three basic mechanisms are known forprotection against pulmonary edema in heartfailure: increased interstitial lymphatic drainage,intralobular interstitial thickening, and pulmonaryvascular remodeling.
In accordance with these mechanisms, PH leadsto a significant reduction in RV output.9 This inturn decreases the blood supply to the pulmonarycapillary bed and protects against edema, in additionto subtly changing the clinical course of the patient:clinical manifestations of pulmonary congestion are reduced to be progressively replaced with systemiccongestion.
In PH due to heart failure, 2 pathophysiologicalaspects should be analyzed: the factors thatcontribute to elevated pulmonary venous pressureand what takes place in the pulmonary vessel for PHto convert from the passive to reactive form.
Factors that Contribute to ElevatingPulmonary Venous Pressure and Maintainingit Elevated
Most patients with chronic heart failure experiencesome degree of PH. However, although many aspectsof this association are not known, it is known thatgreater severity and duration of heart disease isassociated with more severe PH.
The factors that essentially contribute to maintainingchronically high pulmonary venous pressure are leftventricular diastolic dysfunction, mitral valve disease,and left atrial function and remodeling.
In presence of left ventricular dysfunctionand dilation, systolic dysfunction undoubtedlycontributes to elevated pulmonary venous pressure.However, greater severity of diastolic dysfunctionand mitral regurgitation are more closely related tothe development and severity of PH.10
In presence of mitral valve disease, the functionalarea of stenosis and size of the regurgitating orificewhen the valve is incompetent are related to thedegree of venous hypertension. However, theregurgitating or affected orifices are not closelyrelated to the severity of PH and other factors, suchas atrioventricular compliance, intervene in thedevelopment of PH.11
The left ventricle also actively participates in thepathophysiology of PH.12 As has been shown in acanine model of heart failure, the increase in left-ventricular end-diastolic pressure leads to structuralchanges in the atrial wall: hypertrophy of the myocytesand increase in collagen matrix. A positive correlationhas been found between markers of increased atrialcollagen synthesis (greater atrial rigidity) and mPAP.13
This atrial remodeling affects atrial systolic function,but above all, atrial compliance. The greater leftatrial rigidity is transmitted in a retrograde directionwithout damping the high left-ventricular enddiastolic pressure.14
Despite all these aforementioned factors, there isgreat variability in the severity of PH associated withheart failure. Although the mechanisms implicatedin this response variable are not known, it couldbe that genetic factors play a role. In fact, severeelevations in pulmonary artery pressure (systolicpressure ≥80 mm Hg) only occur in less than one-third of patients with chronic elevation of pulmonaryvenous pressure.
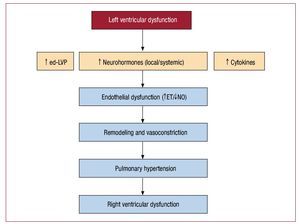
Factors That Contribute to Passive PulmonaryHypertension Becoming Active PulmonaryHypertensionWhile the initial idiopathic pulmonary arteryhypertension (PAH) produces vascular damageof unknown cause, thereby inducing secondaryremodeling and finally PAH, the opposite occursin PH secondary to heart failure: the initial triggeris passive hypertension, which initiates the vascularremodeling process, probably by endothelialdamage, eventually leading to severe and fixed PH(Figure 2).

Figure 2. Possible relationship betweenpresence of left ventricular dysfunctionand secondary events that may lead topulmonary arterial hypertension. ETindicates endothelin; NO, nitric oxide; ed-LVP, end-diastolic left ventricularpressure.
PH associated with chronic heart failure of systolicorigin is the most extensively studied at a molecularlevel. It seems that hemodynamic stress caused bythe passive component of PH, the neurohormonalactivation inherent in heart failure, and local andsystemic production of cytokines trigger endothelialdamage that initiates remodeling in the pulmonaryartery vessel.
Nitric oxide (NO) generated in the endothelialcell of the pulmonary vascular bed acts on smoothmuscle cells causing them to relax, inhibits theirproliferation and hypertrophy and, by means ofa joint action with prostacyclin, inhibits plateletaggregation and adhesion.
Some experimental models and clinical studies ofheart failure point to deficient basal NO productionand suggest that the resulting loss of NO-dependentvasodilation may contribute the development ofPH.15,16
The endothelium (ET) is a vasoactive peptidealso produced by endothelial cells. There are 2 typesof ET receptors: ETA and ETB. The ETA receptorsare localized in smooth muscle cells and mediatevasoconstriction and cell growth. In contrast, ETB receptors are found, above all, in endothelial cellsand stimulation leads to vasodilation throughrelease of NO and prostacyclin. The ETA:ETB ratioin pulmonary arteries is 9:1, and so the net effect ofET release is vasoconstriction and favoring of cellproliferation.
In heart failure, the plasma concentration of ET iselevated and its value closely correlates with mPAPand pulmonary vascular resistance (PVR).17
Both factors dependent on endothelial damage(NO reduction and ET increase) are knownmediators that, by means of vasoconstriction andcell proliferation, initiate vascular remodeling.
Histopathology of Pulmonary HypertensionAssociated With Left Heart Disease
There are few studies that analyze pulmonaryhistopathology in chronic pulmonary venoushypertension, and these are old and limited to mitralstenosis.
Microscopic examination of the pulmonary tissuefrom patients with venous PH shows capillarydistension, thickening, and basement membranerupture and transudation of erythrocytes throughthe damaged membranes to the alveolar spaces.Often, pulmonary hemosiderosis is observed, and this can progress to marked fibrosis. But themost characteristic changes in pulmonary venoushypertension occur in arteries, veins, and lymphvessels (Table 3). The term used to describe thesehistopathological changes is congestive vasculopathyand it has been studied above all in mitral valvedisease.18

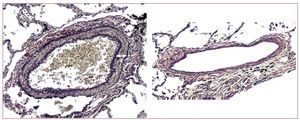
The most characteristic arterial changes occurin the muscular pulmonary arteries. The mostnoteworthy element is medial hypertrophy, whichis often severe and generally more extensive thanobserved in patients with PAH, for comparabledegrees of PH.19
In mitral stenosis, a close correlation has not beenfound between the extent of medial hypertrophy andthe severity of PH.20 However, in advanced systolicheart failure, the extent of medial hypertrophy isrelated to the severity of PH (Figure 3).21

Figure 3. Marked medial hypertrophy of a muscular pulmonary artery in a patient with chronic heart failure (left, arrow indicates the medial thickness),compared to another of similar size with minimal medial thickening (right) in a patient with heart failure but not pulmonary hypertension (van Gieson stain, ×100).
Medial hypertrophy of the muscular arteries isassociated with arteriolar muscularization. Anotherarterial factor often present is intimal fibrosis, whichis generally eccentric and not obstructive.
In veins and venules, medial hypertrophy occursand, with this, arterialization of the venous vesselsand intimal fibrosis. The lymph vessels show markeddilation, with the appearance of lymphangiectasias,particularly when the venous pressure chronicallyexceeds 30 mm Hg.
All these structural changes in the pulmonaryvessels determine whether PH is reactive and fixed orirreversible. The term fixed or irreversible PH meansthat the severity of PH is not reduced with vasodilatordrugs. As such, the term is useful as it represents anapproximation of the severity of PH attributable to vascular remodeling. However, the term irreversibleis not completely correct because once the cause ofvenous hypertension has resolved (for example, afterheart transplantation in chronic heart failure or aftermitral valvuloplasty or valve-replacement surgeryin the case of mitral stenosis), a reverse vascularremodeling process is probably initiated becausepulmonary pressure tends to normalize months oryears after these procedures.22-26
In heart failure with normal left ventricularejection fraction (LVEF), the pathophysiologicaland histopathological aspects have been a lot lesswidely studied. However, in a recent populationstudy in which the prevalence of PH in this type ofpatient was studied by Doppler echocardiography,PH was detected in 83% of patients, and whenpresent, it was often severe. In that same study, itwas found that the severity of PH in many caseswas disproportionate for the degree of left preload,suggesting a reactive vascular component is at work,as in patients with systolic heart failure.27
DIAGNOSIS OF PULMONARYHYPERTENSION IN HEART FAILURE
The definition of PH associated with heart failureis coupled with the need for a hemodynamic studyto confirm that mPAP is ≥25 mm Hg at rest and thatPWP is >15 mm Hg.3
However, hemodynamic study is not necessary inall cases in which PH associated with heart failure isdetected, as echocardiography can provide sufficientinformation for appropriate clinical management.
In systolic heart failure, echocardiographicfindings generally point to an obvious cause of PH.In addition, from tricuspid regurgitation, we canestimate pulmonary systolic pressure and with tissueDoppler studies, using the E/E' ratio, we can obtaina reasonable approximation to the left ventricularfilling pressure. Only when it is necessary toaccurately determine the severity and/or reversibilityof PH (candidate for heart transplantation orventricular support) is hemodynamic study for thispurpose absolutely essential.
However, in a patient with heart failure and normalLVEF, it may be extremely hard to distinguishbetween PH caused by left-ventricular diastolicdysfunction and PAH. The echocardiographicfindings that point to left-ventricular diastolicdysfunction include left ventricular dilation, atrialfibrillation, abnormal left-ventricular filling pattern,and ventricular hypertrophy.28 In this group ofpatients, and although the echocardiographicstudy provides valuable information, sometimescatheterization is necessary to measure PWP orend-diastolic left ventricular pressure to determinewhether they are elevated.28
Nevertheless, even in cases of left-ventriculardiastolic dysfunction, the ventricular filling pressuremay be normal, particularly in patients treatedwith diuretics. Exercise testing or study of volumeoverload has been proposed to uncover "occult" diastolic dysfunction. However, these tools arenot standardized and require better evaluation inorder to be validated.5 On the other hand, in somepatients, it might be difficult to distinguish PAHfrom PH associated with left-ventricular diastolicdysfunction, particularly in those patients with leftventricular preload pressures close to the limit ofnormal (15-18 mm Hg).3
CLINICAL SIGNIFICANCE OF PULMONARYHYPERTENSION IN PATIENTS WITH HEARTFAILURE
In general, PH associated with left-sided heartdisease is a factor resulting in a deterioration infunctional capacity, with worse prognosis and worsesurgical outcomes.
In systolic heart failure, the presence of PH hasimportant functional and prognostic implications.29,30 There is an inverse relationship between peakoxygen uptake (spirometry during exercise testing)and resting pulmonary arterial pressure. In heartfailure, a decrease in systemic vascular resistanceoccurs during exercise while the PVR remains high.Together, this implies that PH contributes to areduction in functional capacity through an increasein RV afterload.31 This hypothesis is based on aclose correlation between peak oxygen uptake andthe right-ventricular ejection fraction at rest andduring exercise.32
In addition, PH in heart failure is closely relatedto inefficient pulmonary ventilation, therebycontributing to exercise-induced hyperpnea anddyspnea in these patients.33,34
But PH in patients with systolic heart failure notonly contributes to their functional deterioration andinfluences the clinical situation, it is also associatedwith worse prognosis and is a variable independentof mortality.30,35 Probably, the negative impact ofPH on survival is due to its impact on RV function,which in turn is an important prognostic marker inadvanced heart disease.36
Less well known is the clinical significance of PHin patients with heart failure and normal LVEF.Recent data from a population-based sampleof 244 patients with heart failure and normalLVEF showed a close relationship between thepresence of PH estimated by echocardiographyand mortality. When this sample was compared toa control group of hypertense patients with heartfailure, the presence of PH had a high predictivevalue for distinguishing between patients with andwithout heart failure. In addition, PH in patientswith heart failure and normal LVEF was theonly echocardiographic variable associated withmortality.27
TREATMENT OF PULMONARYHYPERTENSION IN PATIENTS WITH HEARTFAILURE
There is no specific treatment for PH associatedwith heart failure. Likewise, no drug approved forthe treatment of heart failure is contraindicated dueto the presence of PH.3
In a substantial number of patients, PH associatedwith systolic heart failure is at least partially reversiblewith pharmacological treatment, as the passivecomponent is predominant. Thus, optimizing medicaltreatment (oxygen, diuretics, nitrates, angiotensinconverting enzyme [ACE] inhibitors, angiotensin-IIreceptor antagonists [ARA-II], and b-blockers) andresynchronization therapy significantly reduce PWPand subsequently mPAP.3,37,38
Optimization of medical treatment sometimesrequires a cycle of inotropic treatment (dobutamine,milrinone, or levosimendan).39
However, drugs with a "selective" vasodilatoryeffect in the pulmonary vascular bed and those thatare useful in idiopathic PAH have had a negative orneutral effect on PH secondary to heart failure.
Prostacyclin is very useful in the evaluation ofthe vasoreactivity of the pulmonary vascular bed inheart failure: it reduces PWP and PVR and increasesthe cardiac index. However, chronic administrationby continuous intravenous infusion increasedmortality in treated patients (Flolan InternationalRandomized Survival Trial [FIRST]) by someunknown mechanism.40
Inhalation of NO at doses of 5 to 80 parts permillion (ppm) in moderate or severe heart failurewith associated PH reduces TPG and PVR, butsurprisingly does not reduce pulmonary pressure.Moreover, the decrease in TPG is associated withincreased left-ventricular filling pressure. Perhapsthe selectivity of NO in the pulmonary vascularbed leads to dysfunction in venous return to the leftventricle, and as a result increases the ventricularpreload.41 Thus, inhaled NO is not indicated in themanagement of PH in patients with heart failure.42
However, it has been successfully used to assesspulmonary vasoreactivity,43 as perioperative supportin high-risk valve or coronary artery surgery,44 and forpreventing or treating right ventricular heart failureafter heart transplantation or after implantation ofa left-ventricular support device.45
Sildenafil citrate is a potent selective inhibitor oftype 5 phosphodiesterase (a widespread enzyme inthe pulmonary vascular bed), which induces smoothmuscle cell relaxation and causes vasodilation throughan increase in cyclic GMP. Its acute hemodynamiceffects (oral dose, 50-100 mg) are reduction inmPAP and PVR (to a greater extent than in systemiccirculation) without hardly affecting PWP or thecardiac index. These properties make sildenafil avery useful drug for assessing the vasoreactivity ofpatients with heart failure.46-48 More recent data have shown that an oral dose of 50 mg of sildenafilimproves the functional capacity of patients withsystolic heart failure49 and that chronic treatment for3 and 6 months improves hemodynamics, functionalcapacity, and quality of life in these patients.50,51 Nevertheless, studies are needed that evaluate themechanism of action of sildenafil in heart failure andthat rule out a positive inotropic effect by indirectinhibition of phosphodiesterase 3 (remember that thepositive inotropic effect in heart failure is associatedwith increased mortality), as a step prior to studiesdesigned to assess mortality.52
Selective and nonselective ET receptor antagonists(ETRAs) were shown to improve hemodynamics, ventricular remodeling, and survival in animalmodels of heart failure.53 However, the clinicalresults have been disappointing. The most studiedETRA in PH in patients with heart failure is thenonselective antagonist tezosentan (RandomizedIntravenous Tezosentan [RITZ] 1, 2, 4, and 5 trials).Administered intravenously at a dose of 50-100mg/h, it improves hemodynamics but not clinicalsymptoms or prognosis of acute decompensatedheart failure.54 Perhaps the drug was not been givenat an appropriate dose. Recently it has been foundthat administration of low doses of tezosentan(1-25 mg/h) is hemodynamically active and maylower concentrations of BNP.55 Although thehemodynamic effects with 1 mg/h are minimal andappear late, they may be persistent. New clinicalstudies are necessary to confirm that these doses canlead to clinical improvement.
The second nonselective ETRA studied in chronicheart failure and administered orally is bosentan.In the REACH-1 study, 250 mg of bosentan orplacebo were administered every 12 hours; thestudy was terminated prematurely after findinga high incidence of elevated transaminase in thetreated group.56 On terminating the study, therewere no significant changes in the clinical status ofthe treated and untreated patients. In the ENABLEI/II studies, doses of 125 mg/12 h of bosentan orplacebo were administered to patients with classIII or IV heart failure according to the New YorkHeart Association. After 9 months of follow-up,no differences in clinical status or mortality werefound in the treated group compared to the placebogroup.53
Darusentan, a selective antagonist of ETA receptors, was assessed in the HEAT study, and it wasfound that administration to patients with chronicheart failure improved the cardiac index.57 However,administration at doses of 10 to 300 mg/day for 6months (Endothelin Receptor Antagonist Trial inHeart Failure [EARTH] study) did not improveleft-ventricular remodeling, clinical symptoms, orprognosis.58
Many hypotheses have been proposed to explainthe lack of efficacy of ETRA in patients with heartfailure. One of these is the inappropriate selectionof patients included in clinical trials.2 It may be thatETRA are effective only in a certain subgroup ofpatients with chronic heart failure.59
In view of the above, and at present, none of thepharmacological treatments that have been shownto be effective in PAH are recommended for patientswith systolic heart failure and PH. However, it is veryimportant to continue to research this therapeuticarea, particularly with sildenafil and ETRAs. If PHis a marker of poor prognosis in these patients, itis necessary to continue to search for some other specific effective and safe therapeutic approach,particularly for the subgroup of patients with themost severe forms (disproportionate PH).
In patients with heart failure and normal LVEF,clinical practice guidelines recommend controllingarterial hypertension, prevention of left ventricularhypertrophy or attempting to reverse it through ACEinhibitors or ARA-II, appropriate control of plasmavolume by sodium restriction and diuretics, and,finally, prevention of tachyarrhythmia or controlof heart rate to optimize diastolic filling time with b-blockers or calcium antagonists. In these patients,modest benefit can be obtained in the reductionadmission to hospital by using candersartan.37 But aside from these general recommendations, thereare no data on the influence of specific treatmentsthat have been shown to be effective in PAH fortreatment of heart failure in patients with normalLVEF. Currently, the Evaluating the Effectivenessof Sildenafil at Improving Health Outcomes andExercise Ability in People With Diastolic HeartFailure (RELAX) study is in progress to assess theefficacy of sildenafil in this group of patients.60
MANAGEMENT OF TRANSPLANTATIONCANDIDATES WITH ADVANCED HEARTFAILURE AND PULMONARY HYPERTENSION
This situation is worthy of special mention, asPH associated with heart failure is a risk factor formortality or morbidity after heart transplantation,particularly due to early graft failure associated withRV dysfunction.61-63 And we should bear in mind that early graft failure is the most common cause ofdeath during the first month after transplantation.64
TPG >12 mm Hg and/or precapillary PVR >2.5UW, after vasodilator challenge, are the limitsfor risk above which the risk of death after hearttransplantation increased.65-70 TPG and PVR above these values show a continuous positive correlationwith perioperative mortality, although a limit abovewhich the risk is unacceptable has not be defined.
Although PH is an important risk factor for rightventricular failure after heart transplantation, thereare other factors, some of which are dependent on theRV of donor, which influence perioperative outcomes(hemodynamic management of the donor, ischemicdamage during preservation, and reperfusiondamage after implantation). This would explain whyeven mild PH can have a deleterious influence on theperioperative outcomes and that patients with moresevere PH can undergo transplantation successfully,even with limited hemodynamic repercussions.
In view of the above, in advanced heart failureof the candidate for transplantation, hemodynamicassessment is essential. Before this, it is reasonableto initially optimize the pharmacological treatment for heart failure in accordance with clinicalparameters. When significant PH is detected inclinical examination or Doppler echocardiography,a short course lasting 48 to 72 hours of intravenousinotropic treatment can even be considered prior tothe initial hemodynamic study.
If the pulmonary pressures exceed the limits forrisk in this initial study, study with a vasodilatoris recommended.71 The clinical practice guidelinesof the International Society for Heart and LungTransplantation (ISHLT) recommend conductinga vasodilator challenge if the systolic pulmonaryartery pressure (SPAP) is ≥50 mm Hg and TPGis ≥15 mm Hg or PVRs are >3 UW.72 This is doneusing nitroglycerin, nitroprussiate, prostaglandin E1,prostacyclin, NO, iloprost, and sildenafil. However,no regimen has been shown to be better than anotherand so there are no specific recommendations.3 Likewise, there are no basal hemodynamic parametersthat can predict response to a vasodilator challenge;however, low cardiac index and very high PVR(>6 UW) are predictive of poor response.73
After vasodilator challenge, a large number ofpatients are included in the "reversible" PH categoryand can be put on the waiting list. However, althoughit is a controversial topic, it seems that prognosis afterheart transplantation in this group of patients withreversible PH is slightly unfavorable compared topatients without PH.74 For hemodynamic vigilanceof these patients while they are on the waiting list, it isrecommended to perform right heart catheterizationevery 3 to 6 months.
If the PWP has not decreased below 25 mm Hg andSPAP below 60 mm Hg in the vasodilator challenge,the reversibility of the PH has not been assessed;thus before ruling out heart transplantation for PH,another more effective way of treating the pulmonarycongestion should be sought.72
On the other hand, if the figures for pulmonarypressure after vasodilator challenge lie within therange of reversibility, but they are associated with adrop in systolic artery pressure below 85 mm Hg, therisk is not reduced after transplantation.66
Patients in whom high-risk PH persists aftervasodilator challenge may be candidates for intensivepharmacological inotropic and vasodilator treatment(oral and intravenous), as the PH may finally revertto "reversible" PH. This transformation is what someauthors have denoted "vasodilator conditioning,"75 and in this case a therapeutic escalation of inotropicand vasodilator therapy is necessary underhemodynamic monitoring. There is no well-definedregimen, but a combination of selective inotropicand vasodilator agents are considered. With thisregimen, it is possible to minimize the numberof patients ruled out of heart transplantation for "irreversible" PH.
If high-risk PH persists despite the aboveapproaches, it is possible to add mechanical supportdevices such as intraaortic counterpulsation balloonsand ventricular support devices to pharmacologicaltherapy.72,76
Candidates for transplantation with heartfailure and "reversible" PH who have requiredvasodilator challenge need special management.There are, however, no universally recommendedapproaches although possibilities include 3- or6-monthly reevaluation of the severity of PH whileon the waiting list,72 maintenance of vasodilatortreatment until transplantation,59 appropriate donorselection (rounding up the weight with respect tothe recipient, hemodynamic maintenance with lowdoses or without inotropic agents, and expectedshort ischemia time), and, finally, protection of theright ventricle immediately after surgery with NO,intravenous prostacyclin, iloprost, or sildenafil.
Despite applying all the above options, somepatients do not attain the limits of PH reversibilitythat would allow an acceptable risk for orthotopicheart transplantation. According to the clinicalpractice guidelines of the ISHLT, PVR >5 UWand TPG>16-20 mm Hg, and especially if one ofthe 2 determinations is concurrent with SPAP >60mm Hg after the measures described above should beconsidered a relative contraindication for orthotopicheart transplantation.72
ABBREVIATIONS
mPAP: mean pulmonary artery pressure
PAH: Pulmonary arterial hypertension
PH: pulmonary hypertension
PWP: pulmonary wedge pressure
RV: right ventricular
TPG: transpulmonary gradient
Dr Juan F. Delgado forms part of the of the REDINSCOR cooperativeresearch network, financed by the Ministry of Health and Consumer Affairsthrough the Instituto de Saludo Carlos III.
Correspondence: Dr. J.F. Delgado.
Unidad de Insuficiencia Cardiaca y Trasplante. Servicio de Cardiología.Hospital Universitario 12 de Octubre.
Avda. de Córdoba, s/n. 28041 Madrid. Spain.
E- mail: jdelgado.hdoc@salud.madrid.org