Keywords
INTRODUCTION
For the first time, cardiovascular disease (CVD) exceeds infection and cancer as the leading cause of death throughout most of the world.1 Furthermore, the incidence of CVD at a younger (30-50 years old) age is rising2 as the prevalence of risk factors for CVD (ie hypertension, obesity and type 2 diabetes) increases.3-5 Early atherosclerotic evidence has been found in individuals as young as 18 years of age.6 Compounding this increase is a rise in the number of patients surviving acute cardiovascular insults, which in turn has lead to a concomitant increase in the prevalence of more advanced diseases including heart failure (HF). Cell therapy is a novel technique being applied to the treatment of this continuum of CVDs collectively, with common features and stage specific issues that should be considered. Early in the course of disease, vascular injury is the target; after an infarction, it is damaged myocardium or vasculature; with the onset of heart failure it is the entire tissue or organ (Figure 1).

Figure 1. Tissue integrity reflects a balance between injury and repair. When tissue injury occurs, inflammatory chemo/cytokines are released into the surrounding tissue milieu and circulation to promote recruitment of stem or progenitor cells to the site of injury. If these cells promote repair, inflammation decreases and tissue integrity is restored. However, as progenitor cell numbers decrease or if cells are not functional, repair fails. In this case the balance is tipped toward injury and inflammation increases resulting in negative sequelae of the inflammatory response and tissue injury. Cell therapy should be designed to provide the appropriate cells within an adequate time frame and at a sufficient dose to restore the potential for repair. In the face of chronic disease such at refractory angina or systemic atherosclerotic disease, this may require repeated dosing over months or years. In the case of acute catastrophic injury, this may require a large single dose shortly after injury. When treating end-organ damage, cells alone may not be sufficient and engineered tissues or patches may be required.
WHY CELL THERAPY?
Repair of tissues and organs in the face of normal "wear and tear" occurs throughout much of life but appears to decrease with aging—culminating in the onset of chronic illnesses such as cardiovascular disease. This repair process represents an ongoing balance between tissue injury and repair that is mediated by an exquisite interaction among proand anti-inflammatory signals and endogenous stem or progenitor cells.7 At earlier ages when risk factors are typically lower, enough cells exist to compensate for small insults and maintain repair. With age and chronic disease, failure of repair occurs in part because stem/progenitor cells decrease both in number and in function and can no longer keep pace with demands for repair.7-10 In the face of catastrophic events such as acute trauma, acute myocardial infarction, etc, sufficient numbers of cells either do not exist or cannot be mobilized in time to compensate for the massive injury. In these cases, cell therapy, or delivery of stem/progenitor cells to the site of injury, is a novel approach designed to relieve this failure of endogenous repair.
STEM CELL BIOLOGY AND HEART DISEASE
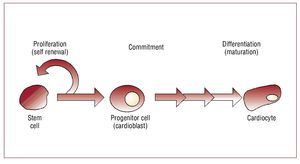
Stem cells are "unspecialized (undifferentiated) master cells"11 that retain the ability to divide and give rise to daughter cells (ie, self-renew) but also to "commit" to become a given type of cell (lineage commitment) and ultimately "differentiate" or mature down that lineage to replace cells that die or are lost (Figure 2). Stem cells typically are defined as pluripotent (can give rise to all cell types in the body), or multipotent (can give rise to multiple cell types but not all) cells that can replicate indefinitely. Progenitor cells are stem cells that have begun to mature in vivo or in vitro, can replicate a limited number of times and typically have less flexibility regarding commitment and maturation. In the blood literature, a stem cell is rigorously defined as a single cell that can replace all of the components of bone marrow and blood. In the regenerative medicine field, this definition has become less clear cut and the terms stem and progenitor cells are often used interchangeably. Typically cell therapy is performed with multipotent stem or progenitor cells (that is, prior to commitment or differentiation) at a stage when they can differentiate down multiple lineages giving rise to different cell types depending on the need. Bone marrow, skeletal muscle, fat, heart, and umbilical cord have all been used as sources of uncommitted adult stem or progenitor cells for cardiovascular cell therapy. Ideally, stem cells could become cardiocytes and integrate with surrounding host tissue. Data from several laboratories suggest this is feasible, although it appears to be a rare event.12 But data further suggest, as shown by Wang et al,13 that when injected into injured or infarcted heart stem cells can develop characteristics of scar. Or in the case of embryonic or human pluripotent stem cells even form benign tumors. Understanding this phenomenon and beginning to control it is an active area of investigation that must be better understood before undifferentiated stem cells per se will be a viable target for clinical myocardial repair.

Figure 2. Stem cell differentiation. Stem cells undergo a process of maturation that involves commitment to a given cell lineage (in this case cardiac) followed by differentiation or maturation down that lineage. Stem cells are capable of self renewal indefinitely whereas progenitor cells have a finite capacity for cell division.
PREVENTING OR REVERSING EARLY CVD (ATHEROSCLEROSIS)
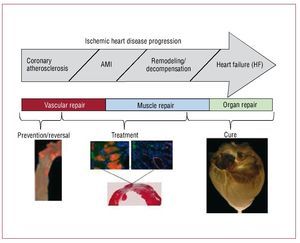
Cell therapy has primarily been used clinically to treat patients after acute myocardial infarction (AMI) or the onset of HF. We propose that earlier assessment and intervention may be warranted. By assessing an individual's capacity for cell-mediated endogenous repair or the failure thereof, it is possible to imagine a unique opportunity for early diagnosis and timely prevention of the consequences of atherosclerosis. Likewise, early cell-based interventions in the face of failed repair could reduce the risk of progression to clinical CVD. These assertions are based on both preclinical and early clinical indications. (see Figure 3 for a proposed disease-based treatment regime).

Figure 3. Cell therapy goals in cardiovascular disease.
In an ApoE knock out mouse model of atherosclerosis, we previously showed that atherosclerotic disease progression is tied to obsolescence of endogenous cells that normally repair and rejuvenate the arteries.10 Furthermore, we demonstrated that repeated treatment with bone marrow-derived progenitor cells (PCs) from young non-atherosclerotic mice prevented atherosclerotic progression in recipients whereas treatment with bone marrow cells from older mice with atherosclerosis was much less effective. In more recent studies, we described sex-based differences in this capacity for endogenous cell-mediated repair.9 In those studies, we determined the capacity for bone marrow mononuclear cells (BM-MNCs) to reverse existing plaque burden after chronic delivery in a sex-matched or sex-mismatched fashion. Only female cells infused intravenously into male ApoE-/- recipients significantly lessened plaque burden. This response correlated with an increase in 3 main cell populations (CD34 CD45, AC133/CD34) in the bone marrow of recipients, but not with total serum cholesterol. The plaque burden reduction was further associated with increases in the Th1-type (pro-inflammatory), decreases in the Th2-type (anti-inflammatory) and shifts in hematopoietic/regulatory cytokines in peripheral blood. This hematopoietic/regulatory and Th2-type cytokine role in the reparative process has not been previously appreciated; but based on these data we hypothesized that cell-mediated vascular repair requires a degree of inflammation to recruit appropriate reparative cells out of the bone marrow resulting in digestion of plaque lesions (presumably by macrophages), engraftment of endothelial progenitor cells (EPCs) and generation of new endothelium. This suggests that a more in-depth understanding of the role of inflammation in atherogenesis is warranted; and that shutting down the inflammatory process in the early phase (by inhibition of Th1-type cytokines either by direct or by nonspecific antagonism) may actually be detrimental to the recruitment of cells necessary for vascular repair. Of note, female bone marrow cells secreted approximately 4 times more granulocyte colony-stimulating factor (G-CSF), a factor known to promote stem cell mobilization out of bone marrow, than did male cells; and the levels of endogenous G-CSF in female ApoE-/- mice were twice those in male mice. These sex-based differences in PC-mediated capacity for vascular repair may not only begin to explain why CVD occurs earlier in men (as repair fails) but may have implications for the use of autologous cells in clinical trials in that female cells far outperformed male cells. In summary, bone marrow derived cells appear to play a role in the maintenance of vascular health. With aging a decrease in specific reparative progenitor cell populations occurs that may begin to account for the accumulation of risk and the rapid progression of atherosclerosis in individuals beyond a certain age. If these continue to be verified clinically, (preliminary data in our hands recapitulates the preclinical findings of differential sex-based cell loss), then cell replacement early in the cardiovascular disease continuum could become a therapeutic option with broad implications for decreasing all aspects of CVD. Further exploration of sex-based differences in stem and progenitor cells present in bone marrow and peripheral blood and in the capacity for cell-mediated disease prevention and treatment is warranted. Although only a small number of patients with advanced atherosclerosis (no AMI or HF) have been studied,14,15 in those patients with refractory angina, BM-MNCs substantially reduced anginal episodes per week to an extent greater than ranolazine, a new anti-anginal agent.16 And the improvement in symptomatology correlated with increased myocardial perfusion. This suggests that a promising future for the use of bone marrow mononuclear cells is in prevention or treatment of early and possibly even asymptomatic CVD.
TREATING ACUTE MYOCARDIAL INJURY
Because the adult myocardium lacks a substantive pool of stem or progenitor cells, it is incapable of effective cardiomyocyte regeneration after a catastrophic injury such as an AMI. In fact, after an AMI, the injured myocardium typically heals to form a non-contracting fibrous scar. If the infarct is large enough, the remaining myocardium ultimately decompensates leading to HF. Current treatment options for acute myocardial infarction and subsequent failure include medical management, cardiac transplantation, mechanical circulatory left ventricular assist devices, or other experimental attempts (artificial hearts), all of which suffer from specific limitations. In light of the limited efficacy and co-morbidities of these current treatment options, alternative, additional long-term therapeutic strategies are needed. Supplying cells capable of cardiac repair has become an emerging therapeutic option (Figure 3).
TRYING TO REBUILD MYOCARDIUM: A CHOICE OF CELL TYPES
After ischemic insult to the myocardium both cardiac muscle and vascular cells die. Thus, repair must encompass not just new cardiac muscle, but vessels capable of supplying oxygen and nutrients to the nascent muscle tissue. Angiogenic and myogenic cells have been investigated both pre-clinically and clinically.
Cardiocytes
At first glance, cardiac myocytes might seem the ideal target for cardiac muscle repair. Yet several major obstacles prevent the use of these cells in vivo. First, for cardiocytes to be used clinically for cell transplantation, large numbers of cells must be readily available. Given the present inability of mature cardiocytes to replicate to a significant degree in vitro or in vivo, this remains unlikely. Second, for cardiocytes to survive, they require a vascular supply far greater than that likely to be obtained in an infarct. Finally, if cardiocytes are to significantly contribute to myocardial function, they must migrate and incorporate into scar, and align in a manner that allows them to electrically and mechanically couple to the remainder of the myocardium. At present, little or no data exist demonstrating this is possible with mature cardiomyocytes.
Skeletal Myoblasts
Adult skeletal muscle contains a population of immature cells capable of repairing damage to the muscle throughout life. These muscle-derived progenitor cells, known as myoblasts, were the first cell type used for functional myocardial repair.17 The advantages of skeletal myoblasts for cardiac repair over cardiac muscle cells include their ability to be grown to large numbers in vitro; their ability to continue to replicate in vivo; and their relative resistance to ischemia, which increases their survival potential in hypoxic regions such as infarcted myocardium. Although myoblasts have been proposed for use relatively early after AMI, their current clinical application is primarily in chronic scarred heart where left ventricle (LV) dysfunction and low ejection fraction (EF) support the need for new muscle formation. They will thus be discussed in more detail later as a therapeutic option for failing heart.
Bone Marrow
Using a bone marrow aspirate that contains a "cocktail" of stem or progenitor cells to accelerate cardiac repair after AMI has gained wide clinical enthusiasm. As a result, more than 1000 patients have been treated with bone marrow cells in phase I and II clinical studies. We have reviewed these individual trials elsewhere18 but the results are summarized here, as are the lessons to be learned. Trials to date have focused on the use of the BMMNCs, endothelial progenitor cells (EPCs), or bone marrow stromal or mesenchymal cells (MSCs) with the most patients being treated following AMI. Three recent meta-analyses suggest that BM-MNCs provide statistically significant yet small (in clinical terms) benefit when administered post-AMI.19-21 However, on close examination of individual studies, the outcomes are discrepant. Whether the discrepancies represent differences in disease context, patient population, cell type and dose, cell manipulation, or some other factor remains to be resolved.
Clinical differences appear to play a role in these discrepancies. For example, BM-MNCs administered in re-perfused and/or stented AMI were not as beneficial. Although a reduction in infarction size was observed, no functional improvement occurred.22 This lack of effect might have been because prompt restoration of coronary flow made cell-based repair unnecessary. This is supported by evidence that when reperfusion and stenting were not uniformly applied, BMMNCs and other bone marrow derived sub-populations (AC133+ EPCs and MSCs) improved myocardial viability, wall motion, coronary flow and left ventricular ejection fraction (LVEF). Yet, all bone marrow fractions were not equally beneficial; in these studies several patients showed either restenosis or de novo lesions after AC133+ EPCs.23 These data suggest that success of bone marrow cells likely lies in the administration of unfractionated cells, permitting cell-cell and cell- tissue interactions in vivo that are otherwise absent when an isolated cell population is administered. In other words, because unfractionated BM-MNCs have both mature and immature progenitors, and as yet undefined cell populations, it is likely that a combination of these cells may be the best choice for the complex types of vascular and cardiac muscle repair demanded after acute myocardial infarction.
Mesenchymal Stem Cells
Bone also contains a population of marrow "stromal" cells that provide support and anchoring for maturing hematopoietic cells. As it became clear these mesenchymal cells had a multipotent potential, a new stem cell population for cardiac repair was born—the bone marrow mesenchymal stem cell. By their very nature, these multipotent stem cells should respond to their microenvironment and develop a corresponding phenotype. This would suggest that, in normal myocardium, bone marrow stem cells could become cardiocytes and integrate with surrounding host tissue. Data from several laboratories suggest this is feasible, although it appears to be a rare event.12 But data further suggest, as shown by Wang et al,13 that when injected into injured or infarcted heart bone marrow mesenchymal stem cells can develop characteristics of scar. Yet recent data reported by Hare et al24 suggest that MSCs offer potential benefit in the AMI setting even after intravenous infusion and minimal accumulation in the myocardium. This has been partially attributed to an anti-inflammatory effect of mesenchymal cells via paracrine influences. Understanding this non-local paracrine effect of cells (and the impact on the inflammatory balance described earlier) and beginning to control it is an active area of investigation that may become the most viable target for myocardial repair.
Other Tissue Derived Cells
Beyond skeletal muscle, bone marrow and peripheral blood, multiple tissues have been suggested as reservoirs for adult stem cells and thus as potential sources for cell isolation for cardiac repair. These include adipose tissue where a mesenchymal cell population can be easily harvested, adult liver that contains significant numbers of regenerative cells, and of course heart.
Cardiac Stem Cells
Currently, at least 4 undifferentiated cell populations (expressing c-kit, MRD-1, isl-1, or sca-1, and lacking expression of hematopoietic markers) have been isolated from neonatal and acutely infarcted failing hearts.25-27 Interestingly, the number of some of these cells was increased after AMI, but was very low in failing hearts suggesting a role of these cells in ongoing repair that becomes insufficient in HF. We isolated an SSEA+ population of uncommitted cardiac progenitor cells (UPCs) in both neonatal and adult rat hearts and showed that these cells can be expanded in vitro, give rise to many of the stem cell populations described previously and be differentiated down cardiac myocyte, smooth muscle and endothelial cell pathways.28 This begins to suggest that adult myocardium contains a population of progenitors that is dynamic and gives rise to a plethora of cell types in vivo including muscle and vascular components. This ability of cardiac-derived stem cells to give rise to what appears to be mature myocardium and vasculature is promising even though the ability to harvest and expand these heart-derived PC populations remains limited—decreasing their current potential for clinical use. Recently, this problem has been partially overcome as Messina et al29 have developed methods to derive myogenic cells from adult cardiac biopsies and we have shown that we can expand SSEA+ cells in vitro to quantities sufficient for cardiac repair.28 The ability to derive, isolate, and expand cardiac stem cells and their potent capacity for cardiac muscle repair suggest a possible role for endogenous mobilization and/or delivery of these cardiac-derived progenitor cells in the future.
TREATING HEART FAILURE
Despite many years of efforts to reduce the pathophysiology of remodeling, up to 50% of post-AMI patients still manifest symptomatic HF by year seven.7 Thus, there is a strong unmet need to develop therapies to improve the quality of life in HF patients while reducing hospitalizations and ultimately mortality. Because remodeling leads to significant fibrosis and loss of contractile cells throughout myocardium and the remaining cells cannot keep pace with the demand for contraction, replacing lost or dying cells with new ones is a therapeutic option that makes sense to both physicians and patients. Having begun in 1998 with the demonstration of LV functional improvement and anti-remodelling effects post-skeletal myoblast (SKMB) transplantation in a rabbit model,17 clinical application of SKMB in approximately 250 patients has been published and was reviewed by us elsewhere.30 The data show a relationship between the contractile impairment at baseline and the LV functional improvement by SKMBs. Specifically, in patients with baseline LVEF <25%, or between 25% and 30% a mean improvement in LVEF was approximately 7%-8%. The only exception was a study by Chachques et al, where mean LVEF improved from 28% to 52% in 14(5) months.31 When the baseline EF was between 30% and 40%, contractility at follow-up improved 9 to 19%. Finally, patients with only mildly abnormal contractility (LVEF >40%), experienced no significant functional benefit. Despite this variable affect on EF, SKMB administration significantly improved patients' HF symptoms—overall by approximately one New York Heart Association functional class—but admittedly the number of controlled studies is small. The first randomized placebo-controlled study (The Myoblast Autologous Grafting in Ischemic Cardiomyopathy Trial) was published in 2008.32 Briefly, it was an international study of 97 HF patients with LVEF ranging from 15% to 35% with a history of AMI and at least 2 contiguous segments with severely compromised function. SKMBs were delivered into non-revascularized myocardium at the time during bypass grafting using 30 injections. The primary endpoints were a change in LVEF at 6 months and LV segmental function recovery. The primary safety endpoints were 30-day and 6-month major adverse cardiac events and arrhythmia rates. Despite no differences in the primary outcomes versus placebo, this study demonstrated several significant points. First, a multicenter trial of SKMB therapy with expanded cells (preserving cell viability) is feasible. Secondly, multiple cell injections into the LV in multiple centers with multiple operators are feasible. Finally, high-dose SKMB transplantation was associated with a significant reduction of LV end-systolic and end-diastolic volumes versus placebo indicating that cell-based treatment exerts anti-remodeling effects of probable long-term importance. These data suggest that in the face of severely damaged myocardium muscle cells can provide a significant functional benefit that surpasses that seen with other cell types to date. Of note is the preclinical evidence that myoblasts in failing myocardium can both form new muscle and importantly recruit or attract nascent vessels to the remuscularized site.
Even though SKMB investigations in HF preceded studies of BM-MNCs in AMI, the progress of SKMB-based therapy has not been equally rapid. This likely reflects both clinical and non-clinical reasons. Clinically, SKMB therapy has been associated with some unique safety and efficacy challenges. As the first cell type used clinically, it was applied in severely-ill unstable HF patients with little understanding of how the cells might work. In those early uncontrolled HF studies arrhythmogenesis was observed in some of the cell recipients.30 Co-administration of amiodarone dramatically reduced the incidence of arrhythmias after SKMB transplantation but did not restore enthusiasm for SKMB studies because of safety issues associated with long-term amiodarone use.18 In this context, SKMBs may suffer from some of the same challenges as BM-MNCs post AMI. Much like with AMI, we do not fully understand the inflammatory milieu of chronic HF. Nor do we appreciate the array of cytokines/chemokines that SKMBs secrete. Borrowing from our atherosclerosis work discussed earlier, we propose that the interaction of the inflammatory milieu of the patient with that of the cells may determine the safety and efficacy of cell therapy. We have demonstrated that the location of cell placement (in relevance to the centre and the periphery of the scar), rather than the cells themselves, may be a critical determinant of safety and efficacy of SKMBs.33 Specifically, a direct injection into the scar center worsened LV performance and caused pro-remodelling effects (increased LV enddiastolic and end-systolic volumes) in a rabbit model. On the contrary, peripheral scar (border zone) administration improved those parameters showing an anti-remodelling benefit. Furthermore, Holter monitoring data showed a higher prevalence of ventricular arrhythmias in animals that received SKMBs into the scar center versus the periphery. These data strongly suggest that the famous real estate slogan "location, location, location" may be in fact applicable to cell therapy with SKMBs in HF.
BM-MNCs have been investigated in HF patients, although to a lesser extent than SKMBs.30 The prominent BM-MNC trials in AMI—Transplantation Of Primary Cells And REcovery of LV Function of Patients with Heart Failure (TOPCARE-HF) and BOne marrOw transfer to enhance ST-elevation infarct regeneration -2 (BOOST-2)—have shown promises that BM-MNCs may be beneficial in this context as well. Albeit in a very small number of patients, targeted placement of BM-MNCs into the ischemic zones resulted in delisting of several patients from transplantation because of increased exercise tolerance.34 This outcome has hinted at possible beneficial effects on BM-MNCs in HF, when placement is targeted. Yet, given the reduced number and migratory capacity of bone marrow derived EPCs seen in patients with advanced CVD and HF,12 it will be interesting to see if BMMNCs are capable of improving LV function in this hostile milieu. Continued clinical studies will definitively answer these questions.
BEYOND CELL TYPES: OTHER ISSUES
Cell therapy trials to date have raised as many questions as they have answered. In both AMI and HF studies, the lessons learned fall into 3 major groups: a) non-standardized protocols have brought discrepant results in similar patient cohorts; b) technical details of cell processing appear to affect outcome; and c) autologous cells have inherent limitations because of the impact of age and disease on their availability and functional proficiency.
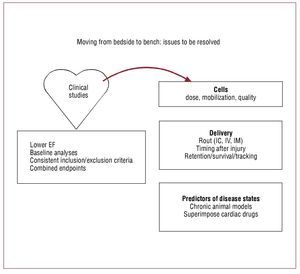
As large-scale multicenter trials proceed, standardization of inclusion and exclusion criteria will largely be resolved. However, several critical questions remain. What are the right endpoints for cell therapy studies? Should we move towards more biologically relevant endpoints recognizing that cells are biological organisms and not chemical substances? Are clinical endpoints or combined endpoints superior to surrogate markers? The success or possible failure of ongoing clinical studies that fail to account for these complexities will unfortunately answer these questions (Figure 4).

Figure 4. Moving from early clinical studies back to the bench: issues to be resolved. EF, ejection fraction; IC, intracoronary; IM, intramyocardial; IV,intravenous.
DOES DELIVERY ROUTE IMPACT THE OUTCOME?
If the above data for skeletal myoblasts is true, delivery route may very well matter. Currently, several routes have been explored. Surgical, or thoracoscopic direct injection into scarred myocardium or more recently endocardial injection remains the preferred routes for failing heart. After AMI the primary route of administration for smaller cells such as the bone marrow mononuclear fraction has been directly into the coronary circulation through the stented or opened vessel. Perhaps the most intriguing route is intravenous (IV) administration. Not only does the IV route make cell delivery in severely ill patients more palatable, but from a scientific perspective it is interesting because IV delivery may alter circulating cytokines/chemokines differently than direct injection of cells into the myocardium, which in turn may play a role both in engraftment of exogenous cells and in the egress of endogenous stem/PCs out of the bone marrow. We have recently obtained preliminary data on the biodistribution of IV administered CD34+ cells that support this supposition. Specifically, we observed an initial cell homing into the liver, the lung and the kidney and a delayed migration into the heart and into the bone marrow. Even though work remains to be done to understand the time course of the cytokine/ chemokine milieu in AMI and HF and the ways it is modified by various cell types, the interactions of bone marrow (and other) cells (ie "cytokine factories") with the inflammatory milieu inherent to the disease may be the primary determinant of both safety and efficacy of cell therapy. At this point, our understanding of these interactions is extremely rudimentary. Centralized collection and storage of bone marrow and blood samples in cell therapy trials should shed light on many of these questions and move us closer to deciphering the mechanisms of endogenous repair and its augmentation by bone marrow cells. This new knowledge will allow us to match cell types, timing of administration and clinical patient characteristics—and perhaps in doing so we can begin to enjoy the full potential of cells for cardiac repair.
CHOOSING THE RIGHT CELL DEPENDING ON STAGE OF DISEASE
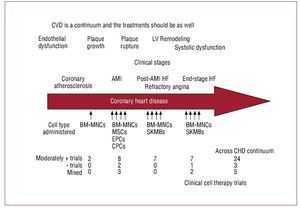
As indicated in Figure 5, multiple populations of cells have been studied clinically throughout the continuum of cardiovascular disease. However, no clear winner has emerged. In fact, despite the differences in cell type and patient populations, a small but quantifiable increase in cardiac function has been observed in many of the studies to date (Figure 5). Yet, it is reasonable to assume that a single cell type is not likely to be sufficient to restore vascular health in atherosclerotic tissue and rebuild integrated functional cardiac muscle in scarred myocardium. Instead, it is likely—as the early evidence indicates—that bone marrow mononuclear cell populations will be capable of vascular repair, whereas more mesenchymal derivatives either from bone marrow, blood, fat or skeletal myoblasts will contribute to muscle regeneration.

Figure 5.Cardiovascular disease represents a continuum of illnesses. Cell therapy should be designed to reflect the underlying injury and tailored accordingly. AMI, acute myocardial infarction; BM-MNC, bone marrow mononuclear cells; CHD, coronary heart disease; CPCs, circulating progenitor cell; CVD, cardiovascular disease; EPCs, endothelial progenitor cells; HF, heart failure; SKMB, skeletal myoblast.
REMAINING CHALLENGES
Several challenges remain if we are to successfully repair infarcted or failing myocardium with any type of cell. The majority of these seem straightforward, but they are complicated by the extreme heterogeneity of cardiovascular disease. For example, the criteria for reproducible engraftment of large numbers of cells may be very different in the early post-myocardial infarction patient versus the patient with end-stage cardiac dysfunction. Similarly, the method of delivering cells (surgical vs endovascular), the concentration of cells delivered, and a host of other criteria ranging from age to co-existing disease states have to be considered as mixed cell transplantation data continues to emerge from clinical studies. Finally, the most significant challenges to cell repair of injured myocardium evolve from our attempts to achieve more than simply halting the progression of cardiovascular deterioration. Pre-clinical data suggest that growth factors, fibroblasts or any number of cells may be able to slow or actually improve diastolic dysfunction.35,36 Yet, if we wish to improve the contractility of the infarcted heart, we must implant cells that can electrically integrate into the heart and survive for extended periods of time. This may involve more than simple cell engraftment, and may ultimately depend on engineered grafts in which we can guarantee nutrient delivery and blood supply in infarcted tissue, and protect the surrounding myocardium from mechanical remodeling and decompensation secondary to these grafts. Although this is not at the forefront of myocardial repair today, it is an area that cannot be ignored.
THE FUTURE: CAN CELL BASED BIO-ARTIFICIAL ORGANS SOLVE THE DONOR SHORTAGE?
The primary goal in the treatment of AMI and HF is restoration of LV function. This can be achieved for a period of time by utilizing medical approaches directed at improving preload and afterload, and by inhibiting signals that govern remodeling of the remaining myocardium— culminating in failure. Transplanting cells into injured heart at least theoretically has the potential of restoring function by directly replacing the cardiac cells that were lost due to injury and disease. However, to be effective, the new cells must engraft in sufficient numbers to either prevent the formation of, or even regenerate cardiac muscle in, fibrotic scar.
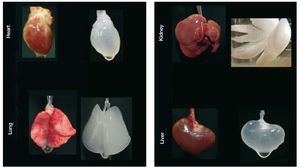
Another approach—folds of magnitude more ambitious—is to bioengineer functional cardiac tissue (Figure 3). This idea of cardiac bioengineering has expanded over the past 10 years. Most studies involve the use of an artificial scaffold (ie hydroxyapatite, collagen, fibrin, etc) seeded with cells and cultivated in the laboratory prior to being used as a cardiac patch. In principle, such a patch could be an on-the-shelf product applied to scar with the goal of re-establishing functional contraction and lessening the risk of aneurysm formation as the patch cells attach and generate contractile force in vivo. The most well-known approach to engineering cardiac patches is the application of neonatal cardiomyocytes to a collagen gel that is then subjected to cyclic mechanical stretch to induce maturation.37,38 Realizing one of the major limitations in cardiac tissue engineering—the difficulty in producing a thick 3-D cardiac tissue due to the high demands for perfusion of cardiac cells—we have undertaken a different approach. Because nature engineered a scaffold that serves as a basis of a heart, we hypothesized that we would be able to wash out the cellular components of the myocardium to obtain a 3-D scaffold comprised of native cardiac extracellular matrix in the original 4-chambered structure of the heart. Then, because the matrix of the major vascular conduits should also remain in place, we should have a perfusable scaffold on which we could reapply cells to build a 3-D cardiac construct or even a whole heart. By recellularizing the decellularized myocardial matrix with the cells that give rise to mature heart, we should be able to generate a myocardial construct that can synchronously contract, respond to drugs and pump against an afterload. We have been able to successfully achieve all these goals and have recently published the characterization of the bioartificial heart construct.39 Briefly, we completely removed cellular structures (<3% of deoxyribonucleic acid remaining) from a rat (or pig) heart using detergent based perfusion decellularization. The cell removal process did not significantly decrease the glycosaminoglycans in the myocardial matrix. Stress strain testing was similar between decellularized rat heart and cadaveric matrices. And the decellularized matrix had a significantly higher tangential modulus than fibrin (a material used by others as a scaffold). The perfusability of the matrix was proven by infusing resin to generate casts showing structural integrity of the vascular tree from the major coronary vessels to fourth/fifth order vessels and by heterotopically transplanting the decellularized construct and showing blood inflow. Recellularization of this matrix was performed by infection of neonatal cells into the matrix in a bioreactor. The construct matured over time, and by 8-10 days, reasonable contractions of the recellularized LV segments were recorded. Histological characterization of the recellularized construct showed live cells expressing myosin heavy chain protein, von Willebrand factor, CD31+ cells, connexin 43, and other markers. Therefore, we have moved one step closer to a creation of autologous organ and constructed a tool to test hypotheses relevant to developmental biology, disease pathophysiology and cell therapy. In doing so, we have, to a reasonable degree, overcome several major limitations of cardiac tissue engineering. Obviously, time will determine which of the current engineering approaches will translate into a clinical product. And although cardiac patches hold promise for a narrow aspect of myocardial repair, building organ constructs via the decellularization-recellularization approach seems to be translatable to other organs (Figure 6) such as kidney, liver, pancreas and lungs that are also often impacted by CVD.

Figure 6. The future of cell therapy may include whole organ bio-engineering. Perfusion decellularized rat organ scaffolds demonstrating the potential of generating scaffolds for complex whole organ bio-engineering.
CONCLUSIONS
Cell therapy has emerged as a new cardiology tool in the past decade. The early clinical phase I and first phase II trials have brought promising results but also offered lessons to be learned going forward. To succeed as a field, it is imperative that we acquire a deeper level of mechanistic understanding of cell-mediated repair throughout the CVD continuum. We propose that the interactions between cells and the specific cytokine milieu during each pathophysiological state from coronary atherosclerosis to end-stage HF will impact the ability of cells to engraft, differentiate and function. We also propose that patients within a temporal window will respond differently depending on their degree of cardiac or vascular damage. As a result, we must more precisely define patient subgroups and the cell types likely to be advantageous in each. This will require understanding the ability of cell populations to alter inflammation, prevent cardiocyte cell death or promote survival, and their capacity to promote angiogenesis and myogenesis both in the acute and chronic settings. By understanding these parameters we should be able to optimize the timing of cell administration so that we can provide sustained anti-remodelling effects and symptomatic relief. Along with cell therapy, cardiovascular tissue engineering is a new frontier in the treatment of CVD. Although the successes are very preliminary, partial and whole bioartificial organ constructs could become a reality and offer a wide range of solutions in the future. Going forward, translating the more recent methods, such as perfusion decellularizationrecellularization, to the bio-organogenesis of other organs impacted by CVD (kidney, lungs, liver) is of crucial importance. Even though we have decades worth of work ahead of us, we cannot forget why we are devoted to finding potential new therapies. Year after year, CVD remains a prominent killer; as the population ages, the need for therapies to decrease cardiovascular morbidity and mortality increases. Achieving that difficult goal will require dedication and cooperation among scientists, clinicians and patients—to move understanding of these new therapies from bench to bedside and if necessary back again.
This section is sponsored by Laboratorio Dr Esteve
Funding and disclosure: This work has been supported in part by the NIH/NHLBI award to D.A.T. (R01-063346), the Minnesota partnership for Biotechnology and Medical Genomics award to Dr Taylor and by funding from the Medtronic Foundation to the Center for Cardiovascular Repair, University of Minnesota. Dr Taylor holds the Medtronic Bakken Chair and directs the Center for Cardiovascular Repair at the University of Minnesota.
Correspondence: Doris A. Taylor,
Director, Center for Cardiovascular Repair University of Minnesota,
312 Church Street SE, Minneapolis MN 55455, USA.