
Thoracic aortic aneurysms are often asymptomatic, remaining undiagnosed until the appearance of catastrophic complications, such as aortic dissection, with very high mortality.1
At present, certain genetic variants are known to predispose individuals to aortic root and ascending aorta disorders, known as hereditary thoracic aortic aneurysm/aortic dissection (HTAAD). Genetic testing (GT) to detect these variants can be very useful for identifying at-risk individuals.2
Traditionally, HTAADs have been considered syndromic (eg, Marfan, Loeys-Dietz, or vascular Ehlers-Danlos syndromes) or nonsyndromic (thoracic aortic aneurysm as an isolated finding). However, the distinction between the two has become less clear, in view of the wide phenotypic variability seen in many of the variants discovered.
We believe that an outpatient office specifically for inherited aortopathies (IA) would help identify these patients more effectively by implementing GT in a more complete and efficient manner.
This single-center retrospective analysis included patients studied in the IA outpatient office of a tertiary hospital between 2010 and 2020. The first visit included a thorough physical examination to look for the clinical signs classically associated with syndromic HTAAD plus a transthoracic echocardiogram to obtain aortic root and ascending aorta measurements, as per current recommendations.3 Afterwards, blood or saliva samples were collected for GT from patients who met the following criteria: body size- and age-adjusted aortic diameters above normality (Z-score> 2) in the absence of other risk factors; increased diameters along with other phenotypic traits classically associated with syndromic HTAAD or a family history of aortic events or sudden cardiac death; or first-degree relatives of patients with any of the above characteristics and/or who have a known genetic variant possibly associated with an aortopathy. GT was performed by combining massive sequencing (NGS) and Sanger methods. The first patient from a family to visit the outpatient office was considered the index case. When a genetic variant with a certain probability of being pathogenic was found, cascade screening was initiated, offering GT to all first-degree relatives. Screening was not performed in patients who declined or whose first-degree relationship was uncertain. The study was approved by the local ethics committee, and patients signed written informed consent before samples were collected for GT.
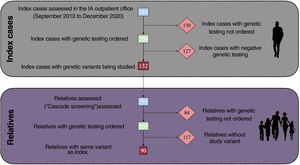
After evaluating 389 index patients, targeted GT was ordered for 259 (66.6%). A genetic variant of interest was obtained in 132 (50.9%) (figure 1), 46 of which were pathogenic or likely pathogenic (17.7%). Based on these findings, 291 relatives were evaluated and GT was subsequently ordered for 207 (71.1%). In this latter group, 90 (43.5%) were positive for the variant studied and 117 were negative. Patients who had variants, whether pathogenic or not, and patients with negative GT but persistent suspicion of a predisposition to aortopathy received regular follow-up.

In all, 140 patients were carriers of FBN1 variants, 11 had variants in genes coding for TGF-B or its receptors, 19 in COL3A1, 22 in other collagen-forming proteins, and 22 in genes coding for vascular smooth muscle cell proteins. The characteristics of these patients plus the clinical characteristics of carrier patients are listed in table 1.
Characteristics of patients with suspicion of hereditary thoracic aortic aneurysms and dissections and finding of a variant of interest in genetic testing
| Total (N=222) | Variants in FBN1 (n=140) | Variants in TGFBR1, TGFBR2, or TGB3 (n=11) | Variants in Col3A1 (n=19) | Variants in other collagen-coding genes (1A1, 1A2, 5A1) (n=22) | Variants in other VSMC protein genes (ACTA, JAG, etc.) (n=22) | |
|---|---|---|---|---|---|---|
| Syndrome diagnostic criteria | ||||||
| Marfan syndrome | 125 (56.3) | 125 (89.3) | — | — | — | — |
| Loeys-Dietz syndrome | 10 (4.5) | — | 10 (90.9) | — | — | — |
| Vascular Ehlers-Danlos syndrome | 14 (0.6) | — | — | 14 (73.7) | — | — |
| Index cases | 132 (59.5) | 86 (61.4) | 6 (54.5) | 8 (42.1) | 12 (54.5) | 12 (54.5) |
| Carrier relatives | 90 (40.5) | 54 (38.6) | 5 (45.5) | 11 (57.9) | 10 (45.5) | 10 (45.5) |
| Age, y | 34.8±18.1 | 32.7±17.4 | 31.0±19.1 | 39.1±18.9 | 34.1±18.1 | 45.2±14.9 |
| Men | 118 (53.2) | 72 (51.4) | 7 (63.6) | 8 (42.1) | 13 (59.1) | 15 (68.2) |
| Ascending aorta | ||||||
| Mild aortic dilatation | 105 (47.3) | 79 (56.4) | 1 (9.1) | 7 (36.8) | 7 (31.8) | 6 (27.3) |
| Ascending thoracic aortic aneurysm | 39 (17.6) | 26 (18.6) | 5 (45.5) | 0 | 2 (9.1) | 5 (22.7) |
| Type A dissection | 13 (5.9) | 11 (7.9) | 0 | 0 | 0 | 2 (9.1) |
| Aortic rupture | 1 (0.5) | 0 | 1 (5.3) | 0 | 0 | 0 |
| Surgical procedure | 48 (21.6) | 34 (24.3) | 4 (36.4) | 0 | 2 (9.1) | 7 (31.8) |
| Bono-Bental | 20 (41.7) | 14 (41.5) | 1 (25) | 0 | 0 | 5 (71.4) |
| Yacoub | 15 (31.3) | 11 (32.4) | 2 (50) | 0 | 0 | 2 (9.1) |
| David | 11 (22.9) | 9 (26.5) | 1 (25) | 0 | 0 | 1 (14.3) |
| Aneurysms in other locations | 7 (3.2) | 0 | 2 (10.6) | 2 (10.5) | 2 (9.1) | 0 |
| Bicuspid aortic valve | 4 (1.8) | 2 (1.4) | 0 | 0 | 0 | 1 (4.5) |
| Mitral valve prolapse | 59 (26.6) | 48 (34.3) | 1 (9.1) | 7 (36.8) | 2 (9.1) | 0 |
| Ectopia lentis | 46 (320.7) | 46 (32.9) | 0 | 0 | 0 | 0 |
| Characteristics of genetic variants | ||||||
| Pathogenic/likely pathogenic* | 146 (65.7) | 121 (86.4) | 9 (81.8) | 12 (63.2) | 0 | 1 (4.5) |
| Possibly pathogenic | 6 (2.7) | 1 (0.7) | 1 (9.1) | 0 | 0 | 4 (18.2) |
| VUS | 70 (31.5) | 18 (12.9) | 1 (9.1) | 7 (36.8) | 22 (100) | 17 (77.3) |
| Previously described in the literature | 76 (34.2) | 66 (47.1) | 4 (36.4) | 1 (5.3) | 0 | 4 (18.2) |
*The genetic variants identified were chosen following the recommendations of the Human Genome Variation Society (HGVS) and the American College of Medical Genetics (ACMG). In the specific case of FBN1, variants were classified as pathogenic if they met the criteria for causal FBN1 mutations according to the modified Gante criteria.
Qualitative variables are expressed as No. (%) and quantitative variables as mean±standard deviation.
In short, more than 50% of the tests yielded a variant of interest. Renner et al.4 found variants in 40.7% of 199 individuals with clinical characteristics of syndromic or nonsyndromic HTAAD. Conversely, a series of unselected patients with only a history of thoracic aortic aneurysm or dissection reported 3.9% to 5% positive reactions for pathogenic or likely pathogenic variants and around 25% when including variants of uncertain significance (VUS).5 Hence, GT requests should be based on clinically guided criteria. In our study, 90 relatives were found to have the study variants, which could have direct implications for follow-up, medical treatment, prophylactic surgical repair, and family planning.
The highest percentage of pathogenic or likely pathogenic variants was obtained in FBN1, a gene associated with Marfan syndrome, the most thoroughly studied IA. Follow-up of patients with a VUS will be of particular interest, as it could shed light on the possible prognostic significance of these variants. Initiatives such as the Spanish Network for Genetic Aortic Pathologies (REPAG) would undoubtedly help reach this goal.
In summary, an outpatient office specifically for IA could help guide individualized treatment for these patients, as well as make sense of the immense amount of information obtained from GT.
FUNDINGNo funding was received to perform this study.
AUTHORS’ CONTRIBUTIONSV.M. Becerra-Muñoz performed the analyses and wrote the manuscript; V.M. Becerra-Muñoz, A. Díaz-Expósito, and V. Doncel-Abad collected the clinical and genetic data; P. Fernández-García, J.L. López-Benítez, and F. Cabrera-Bueno attended patients in the inherited aortopathy outpatient office; F. Cabrera-Bueno is the coordinator of the inherited aortopathy unit; and V.M. Becerra-Muñoz, A. Díaz-Expósito, V. Doncel-Abad, P. Fernández-García, J.L. López-Benítez, and F. Cabrera-Bueno reviewed and corrected the final content of the text.
CONFLICTS OF INTERESTThe authors declare that they have no conflicts of interests related to this study.