Recent advances in genetics have led to the discovery of new genes associated with pulmonary arterial hypertension, such as TBX4 and KCNK3. The phenotype and prognosis associated with these new genes have been scarcely described and their role in the Spanish population is unknown. The aim of this study was to characterize the genetics of a Spanish cohort of patients with idiopathic and hereditary pulmonary arterial hypertension and to describe the phenotype and prognostic factors associated with BMPR2 and the new genes (KCNK3 and TBX4).
MethodsA total of 165 adult patients were screened for BMPR2, KCNK3, and TBX4 mutations, 143 with idiopathic pulmonary arterial hypertension and 22 with hereditary pulmonary arterial hypertension. Baseline characteristics and survival were compared among the different subgroups and predictors of poor outcomes were analyzed. We also performed family screening.
ResultsThe genetic study identified a possibly associated mutation in 11.10% of the idiopathic cases (n = 16) and in 68.18% of the hereditary cases (n = 15). There were 19 mutations in BMPR2, 4 in TBX4, and 3 in KCNK3. The forms associated with TBX4 showed the highest survival rate (P < .01). Advanced functional class at diagnosis was the only factor associated with poor outcomes in the hereditary forms. In the family screening, 37.5% of relatives tested positive.
ConclusionsThe genetics of pulmonary arterial hypertension in the Spanish population may differ from other populations, with a lower proportion of BMPR2 causative mutations. In our cohort, TBX4-related forms of pulmonary arterial hypertension showed a more benign course and late diagnosis was the only predictor of adverse outcomes in the hereditary forms of the disease.
Keywords
Pulmonary arterial hypertension (PAH) is an uncommon disease associated with a poor prognosis in untreated individuals. Its forms of presentation are diverse and include idiopathic PAH (IPAH), which has no known cause, and hereditary PAH (HPAH), which is related to a genetic alteration or shows familial aggregation.
Idiopathic PAH is a rare disease affecting 5.6 adults/million population in Spain, with 1.2 new cases per million population a year according to data from the Registro Español de Hipertensión Arterial Pulmonar (REHAP), a voluntary Spanish registry of patients with PAH founded in 2007. The methodology of this registry has been detailed elsewhere.1
The first gene linked to the development of HPAH was BMPR2.2 This gene encodes the bone morphogenetic protein receptor type 2 and regulates multiple cellular functions. Mutations in BMPR2 have been described in 75% of the hereditary forms of PAH and 25% of the idiopathic forms. These mutations show an autosomal dominant inheritance pattern, incomplete penetrance (∼20%) varying by sex (42% in women vs 14% in men),2,3 and variable expressivity.4–7 The underlying molecular mechanisms are still unknown.8–12
There are more than 300 known mutations in BMPR2 spread throughout its 13 exons and 4 protein domains.4,5 Most of these mutations (70%) are frameshifts, nonsense mutations, or deletions4; the remainder (30%) are missense mutations.
Recent advances in genetics, such as massively parallel sequencing, have enabled the discovery of new genes related to HAP,4,13–20 such as KCNK3, encoding for a pH-dependent potassium channel,13TBX4, encoding for the TBX4 transcription factor14 involved in embryonic development and related to small patella syndrome, and the CAV1,15TOPBP1, SMADS, and NOTCH34,13–20 genes. In addition, EIF2AK4 has been linked to the development of recessive forms of pulmonary veno-occlusive disease.16,17 Thus, PAH is a genetically complex disease with a growing number of implicated genes, although there is little understanding of the phenotype associated with each gene.
The objectives of this study were to genetically and phenotypically characterize a Spanish cohort of IPAH and HPAH patients and analyze the prognostic impact associated with the genetic variants, the predictors of death or placement on the lung transplant list, and the familial impact in terms of penetrance and family screening dynamics.
METHODSThis multicenter Spanish study of PAH genetics was begun in November 2011. It involved an observational and ambispective study of adult IPAH/HPAH patients recruited in 2 Spanish centers (Hospital Universitario 12 de Octubre, Madrid, and Hospital Universitario Vall d’Hebron, Barcelona, and included in the Biobanc tissue bank of the Hospital Clínic de Barcelona). Patients with veno-occlusive disease were excluded from the present analysis. The study comprised 2 cohorts: a cohort of consecutive patients treated in the Hospital Universitario 12 de Octubre and Hospital Universitario Vall d’Hebron (January 2011-May 2015) and a cohort of patients included in the Biobanc tissue bank of the Hospital Clínic de Barcelona (January 2013-March 2014). The diagnosis of PAH was made according to international recommendations.21 Prior to the genetic analysis and based on the presence of a family history of PAH, the patients were classified as having HPAH (positive family history) or IPAH (negative family history). After the genetic analysis, they were reclassified after the genetic analysis as having idiopathic forms (negative family history and without genetic alterations) and hereditary forms (patients with a positive family history and positive genetic test results, patients with a positive family history and negative genetic test results, and patients with a negative family history but positive test results). Clinical data were obtained from REHAP and the genetic testing was performed in the Institute of Medical and Molecular Genetics (INGEMM) of the Hospital Universitario La Paz, Madrid.
Clinical CharacterizationThe following data were collected in all patients at diagnosis: demographic characteristics (age and sex), clinical features (New York Heart Association [NYHA] functional class, 6-minute walk test, syncope), hemodynamic variables (right atrial pressure, mean pulmonary artery pressure, cardiac index and arterial pulmonary vascular resistance, acute vasodilator test response), and pulmonary function parameters (forced expiratory flow in 1 s, forced vital capacity, and carbon monoxide diffusing capacity). Clinical and radiological screening for small patella syndrome was performed in carriers of the TBX4 mutation.
Molecular StudySanger sequencing was performed of all exon and exon-intron boundaries of BMPR2, KCNK3, and TBX4. Screening for structural variants and chromosome rearrangements was performed using MLPA (multiplex ligation-dependent probe amplification) ( of the ).
The variants identified were classified according to their frequency in the general population. Variants absent from public databases (Exome Variant Server, 1000G, and of the ) or with very low allele frequencies (< 0.01%) were considered mutations. The pathogenicity of variants was assessed by taking into account their effect on the protein level, their presence in previous reports in the literature, the evidence of cosegregation with the disease, and the in silico predicted impact on the protein using widely used software (Polyphen2, MutationTaster of the , SIFT [Sorting Intolerant from Tolerant], MutPred, SNPs&GO). The variants identified were classified as pathogenic, possibly pathogenic, variants of unknown significance, and nonpathogenic variants ( of the ). Once the proband study was completed, a genetic study of the first-degree relatives of the mutation carriers was performed and the genealogical tree was constructed.
Statistical AnalysisThe statistical software Stata (version 13, Stata Corp.; College Station, Texas, United States) and IBM SPSS 22 (SPSS, Inc.; Chicago, Illinois, United States) were used for the statistical analysis and the data are presented as mean ± standard deviation with a level of significance of P ≤ .05. Kaplan-Meier univariate survival analysis used the time of diagnosis as the start of follow-up and the 15th June 2015 as the end of follow-up, with a maximum follow-up of 15 years (generalized type I censoring). The log rank test was used to compare the survival rates of the IPAH and HPAH patients and the forms associated with the different genes; outcome was defined as death or placement on the lung transplant waiting list. The hazard ratio and 95% confidence interval were calculated for classical prognostic factors (right atrial pressure, cardiac index, 6-minute walk test, NYHA functional class at diagnosis, sex, mean pulmonary artery pressure, and pulmonary vascular resistance), as well as hereditary disease. Also evaluated was the interaction of the classical prognostic factors with hereditary disease. Those variables showing a significance level of P < .10 in the univariate analysis were included in the multivariate Cox regression model, using forward and backward stepwise selection to confirm the results.
The study was performed in accordance with the principles of the Declaration of Helsinki and Spanish law on data protection and was approved by the ethics committees of the participating centers. All patients provided informed consent.
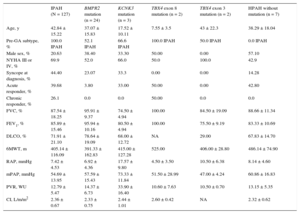
RESULTSOf the 165 patients diagnosed with PAH between November 1st 2011 and May 1st 2015; 143 (86.6%) had IPAH and 22 (13.3%) had HPAH. The baseline characteristics of these patients are shown in Table 1.
Baseline Characteristics of the Population. Comparison of Hereditary Pulmonary Arterial Hypertension and Idiopathic Pulmonary Arterial Hypertension
| Total population (n = 165) | HPAH (n = 38) | IPAH (n = 127) | P | |
|---|---|---|---|---|
| Pre-GA subtype↕ | 143 IPAH, 22 HPAH | 16 IPAH, 22 HPAH | 127 IPAH | — |
| Age, y | 40.68 ± 16.13 | 35.18 ± 17.51 | 42.84 ± 15.22 | .006 |
| Male sex, % | 24.8 | 38.4 | 20.63 | .02 |
| NYHA, % | .054 | |||
| I-II | 33.4 | 43.6 | 30.1 | |
| III | 61.8 | 43.6 | 66.6 | |
| IV | 4.2 | 8.1 | 3.1 | |
| Syncope at diagnosis, % | 38.2 | 17.9 | 44.4 | .003 |
| Acute responder, % | 33.30 | 12.82 | 39.68 | .001 |
| Chronic responder, % | 20.60 | 2.63 | 26.10 | .005 |
| FVC, % | 88.89 ± 16.9 | 93.67 ± 11.35 | 87.54 ± 18.25 | .03 |
| FEV1, % | 87.29 ± 14.89 | 92.44 ± 12.1 | 85.89 ± 15.46 | .01 |
| DLCO, % | 72.23 ± 20.67 | 76.00 ± 24.14 | 71.91 ± 21.10 | NS |
| 6MWT, m | 410.61 ± 116.76 | 417.19 ± 106.64 | 405.14 ± 116.09 | NS |
| RAP, mmHg | 7.59 ± 4.79 | 8.16 ± 5.58 | 7.42 ± 4.53 | NS |
| mPAP, mmHg) | 55.77 ± 14.43 | 59.28 ± 15.54 | 54.69 ± 13.95 | NS |
| CI (L/m/m2) | 2.34 ± 0.72 | 2.31 ± 0.67 | 2.36 ± 0.67 | NS |
| PVR, WU | 13.67 ± 10.93 | 15.57 ± 9.03 | 12.79 ± 5.47 | .02 |
6MWT, 6-minute walk test; CI, cardiac index; DLCO, diffusing capacity of carbon monoxide; FEV1, forced expiratory volume in the first second; FVC, forced vital capacity; HPAH, hereditary pulmonary arterial hypertension; IPAH, idiopathic pulmonary arterial hypertension; NS, not significant; NYHA, New York Heart Association; mPAP, mean pulmonary artery pressure; Pre-GA subtype, PAH subtype before genetic analysis; PVR, pulmonary vascular resistance; RAP, right atrial pressure.
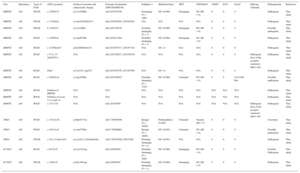
A positive genetic test was observed in 31 probands (18.78%). Specifically, in 16 patients with IPAH (11.10%) and in 15 patients with HPAH (68.18%). Nineteen BMPR2 variants ( of the ) were identified in 24 probands, as well as 3 TBX4 variants in 4 probands and 2 KCNK3 variants in 3 probands ( of the ). The baseline clinical characteristics according to the affected gene are shown in Table 2.
Baseline Characteristics According to the Affected Gene
| IPAH (N = 127) | BMPR2 mutation (n = 24) | KCNK3 mutation (n = 3) | TBX4 exon 8 mutation (n = 2) | TBX4 exon 3 mutation (n = 2) | HPAH without mutation (n = 7) | |
|---|---|---|---|---|---|---|
| Age, y | 42.84 ± 15.22 | 37.07 ± 15.83 | 17.52 ± 10.11 | 7.55 ± 3.5 | 43 ± 22.3 | 38.29 ± 18.04 |
| Pre-GA subtype, % | 100.0 IPAH | 52.1 IPAH | 66.6 IPAH | 100.0 IPAH | 50.0 IPAH | 0.0 IPAH |
| Male sex, % | 20.63 | 38.40 | 33.30 | 50.00 | 0.00 | 57.10 |
| NYHA III or IV, % | 69.9 | 52.0 | 66.0 | 50.0 | 100.0 | 42.9 |
| Syncope at diagnosis, % | 44.40 | 23.07 | 33.3 | 0.00 | 0.00 | 14.28 |
| Acute responder, % | 39.68 | 3.80 | 33.00 | 50.00 | 0.00 | 42.80 |
| Chronic responder, % | 26.1 | 0.0 | 0.0 | 50.00 | 0.0 | 0.0 |
| FVC, % | 87.54 ± 18.25 | 95.91 ± 9.37 | 74.50 ± 4.94 | 100.00 | 84.50 ± 19.09 | 88.66 ± 11.34 |
| FEV1, % | 85.89 ± 15.46 | 95.94 ± 10.16 | 80.50 ± 4.94 | 100.00 | 75.50 ± 9.19 | 83.33 ± 10.69 |
| DLCO, % | 71.91 ± 21.10 | 78.64 ± 19.09 | 68.00 ± 12.72 | NA | 29.00 | 67.83 ± 14.70 |
| 6MWT, m | 405.14 ± 116.09 | 391.33 ± 162.83 | 415.00 ± 127.28 | 525.00 | 406.00 ± 28.80 | 486.14 ± 74.90 |
| RAP, mmHg | 7.42 ± 4.53 | 6.92 ± 4.36 | 17.57 ± 9.80 | 4.50 ± 3.50 | 10.50 ± 6.38 | 8.14 ± 4.60 |
| mPAP, mmHg | 54.69 ± 13.95 | 57.59 ± 15.43 | 73.33 ± 11.84 | 51.50 ± 28.99 | 47.00 ± 4.24 | 60.86 ± 16.83 |
| PVR, WU | 12.79 ± 5.47 | 14.37 ± 6.73 | 33.90 ± 16.40 | 10.60 ± 7.63 | 10.50 ± 0.70 | 13.15 ± 5.35 |
| CI, L/m/m2 | 2.36 ± 0.67 | 2.33 ± 0.75 | 2.44 ± 1.01 | 2.60 ± 0.42 | NA | 2.32 ± 0.62 |
6MWT, 6-minute walk test; CI, cardiac index; DLCO, diffusing capacity of carbon monoxide; FEV1, forced expiratory volume in 1 second; FVC, forced viral capacity; HPAH, hereditary pulmonary arterial hypertension; IPAH, idiopathic pulmonary arterial hypertension; MUT, mutation; NA, not available; NYHA, New York Heart Association; mPAP, mean pulmonary artery pressure; Pre-GA subtype, pulmonary arterial hypertension subtype before genetic analysis; PVR, pulmonary vascular resistance; RAP, right atrial pressure.
The genetic study revealed a BMPR2 variant in 9.1% of the IPAH patients and 50.0% of the HPAH patients. The 19 BMPR2 variants (8 missense mutations) were found in 24 probands from 20 unrelated families, with 2 unrelated probands showing the same variant (p.Asp491Glu) ( of the ). Eight of these variants have been described in the literature. Of the other 11, 9 were considered pathogenic: 7 involved radical changes (frameshifts, structural variants, or intronic variants with probable effects on splicing) and 2 were located in residues previously described to be mutated in patients with PAH ((p.Cys420Phe located in the same codon as p.Cys420Tyr of the and p.Cys420Arg of the , and p.As487His located in the same codon as p.Asp487Val of the ).). The remaining 2 (p.Cys34Phe and p.Arg365His) were considered possibly pathogenic due to the results of the bioinformatic prediction software. Cosegregation was only demonstrated in 1 family (p.Asn442ThrfsX31). Cosegregation could not be ruled out for the other variants but could not be confirmed given the incomplete penetrance associated with BMPR2 mutations (Table 3). The baseline characteristics of the patients according to the type of mutation are shown in of the .
Results of the Bioinformatics Study of the Pathogenicity of Previously Undescribed Variants
| Gen | Inheritance | Type of PAH | cDNA position | Position in protein and aminoacidic change | Genomic localization (GRCH38/HG38) | Poliphen-2 | MutationTaster | SIFT | SNPs&GO | 1000G | EVS | ExAC | Splicing (Alamut) | Pathogenicity | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BMPR2 | AD | FPAH | c.1259G>T | p.Cys420Phe | chr2:203397438 | Damaging (P = .990) | DC (0.999) | Damaging | DC (RI = 9) | 0 | 0 | 0 | Pathogenic | This study | |
| BMPR2 | AD | FPAH | c.1325delA | p.Asn442ThrfsX31 | chr2:203407082_203407082 | N/A | N/A | N/A | N/A | 0 | 0 | 0 | Pathogenic | This study | |
| BMPR2 | AD | FPAH | C.101G>T | p.Cys34Phe | chr2:203329556 | Probably damaging (P = 1) | DC (0.999) | Damaging | DC (RI = 8) | 0 | 0 | 0 | Possibly pathogenic | This study | |
| BMPR2 | AD | IPAH | c.1459G>C | p.Asp487His | chr2:203417484 | Probably damaging (P = 1) | DC (0.999) | Damaging | DC (RI = 8) | 0 | 0 | 0 | Pathogenic | This study | |
| BMPR2 | AD | IPAH | c.1138DelAT | p.Ile380GlnfsX18 | chr2:203397317_203397318 | N/A | DC (1) | N/A | N/A | 0 | 0 | 0 | Pathogenic | This study | |
| BMPR2 | AD | IPAH | c.77-5_77-2delTTTA | N/A | chr2:203329527_203329530 | N/A | N/A | N/A | N/A | 0 | 0 | 0 | Pathogenic (loss of the acceptor canonical splice site) | Pathogenic | This study |
| BMPR2 | AD | IPAH | Dup* | p.Tyr218_Arg225 | chr2:203383576_203383599 | N/A | DC (1) | N/A | N/A | 0 | 0 | 0 | Pathogenic | This study | |
| BMPR2 | AD | IPAH | c.1094G>A | p.Arg365His | chr2:203395643 | Probably damaging (P = .529) | DC (0.999) | Tolerated | DC (RI = 0) | 0 | 0 | 2/121296 Het | Possibly pathogenic | This study | |
| BMPR2 | AD | IPAH | Deletion of BMPR2 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | Pathogenic | This study | |
| BMPR2 | AD | IPAH | Deletion of exons 11,12 and 14 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | Pathogenic | This study | |
| BMPR2 | AD | IPAH | c.1277-3G | N/A | chr2:20340700 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | Pathogenic (loss of the acceptor canonical splice site) | Pathogenic | This study |
| TBX4 | AD | IPAH | c.1351A>G | p.Met451Val | chr17:59560590 | Benign (P < .0005) | Polimorphism (0.888) | Tolerated | Neutral (RI = 7) | 0 | 0 | 0 | Uncertain | This study | |
| TBX4 | AD | IPAH | c.1423A>C | p.Asn475His | chr17:59560662 | Benign (P < .0005) | DC (0.995) | Tolerated | DC (RI = 0) | 0 | 0 | 0 | Possibly pathogenic | This study | |
| TBX4 | AD | FPAH | c.310_312insAAG | p.Lys103_Val104insGlu | chr17:59543208_59543209 | Probably damaging (P = 1) | DC (0.991) | N/A | N/A | 0 | 0 | 0 | Pathogenic | This study | |
| KCNK3 | AD | IPAH | c.641T>G | p.Leu214Arg | chr2:26950892 | Probably damaging (P = .998) | DC (0.999) | Damaging | DC (RI = 9) | 0 | 0 | 0 | Possibly Pathogenic | This study | |
| KCNK3 | AR | FPAH | c.316G>C | p.Gly106Arg | chr2:26950567 | Probably damaging (P = 1) | DC (0.999) | Damaging | DC (RI = 9) | 0 | 0 | 0 | Pathogenic | This study |
AD, autosomal dominant inheritance; AR, autosomal recessive inheritance; DC, disease causing; Dup*, duplication c.653_676dupATAAAGGCTCCTTGGATGAGCG; FPAH, familial pulmonary arterial hypertension; IPAH, idiopathic pulmonary arterial hypertension; N/A, not applicable; RI, confidence scale; SIFT, Sorting Intolerant from Tolerant.
Various predictors of pathogenicity with the following scales were applied: Polyphen-2 (its scale ranges from 0 to 1: values closer to 1 have a higher probability of being classified as pathogenic [“damaging” in the Table]); MutationTaster (its scale also ranges from 0 to 1 and values close to 1 are correlated with a higher possibility of pathogenicity); SIFT (this program classifies variants as “damaging” if they are possibly pathogenic and “tolerated” if they are benign); SNPs&GO (it calculates the possibility of pathogenicity with a confidence scale of prediction ranging from 1 to 9, with 1 indicating lowest confidence and 9 indicating maximum confidence). In addition, these variants were analyzed in various control populations, as shown in the Table: EVS (Exome Variant Server), 1000G (1000 Genomes database), and ExAc (Exome Aggregation Consortium). Values equal to 0 indicate that no clear change is found in that database; for those that do show a change, the allelic content is shown with respect to the total.
Transcript used to analyze TBX4: ENST00000240335. Transcript used to analyze BMPR2: ENST00000374580. Transcript used to analyze KCNK3: ENST00000302909.
Two novel missense mutations (p.Leu214Arg and p.Gly106Arg) were identified in exon 2 of KCNK3 in a patient with IPAH and 2 related patients with HPAH. p.Gly106Arg was identified in a homozygous state in a proband with an aggressive form of PAH and was considered pathogenic given its cosegregation with the disease. p.Leu214Arg was classified as possibly pathogenic and its in silico analysis suggested a probable pathogenic effect (Table 3 and of the ).
Mutations in TBX4Two missense mutations (p.Met451Val and p.Asn475His) were detected in exon 8 of TBX4 in 2 patients with IPAH. A 3-nucleotide insertion in exon 3 was found in 2 related patients with HPAH. The latter variant was considered pathogenic. p.Asn475His was considered possibly pathogenic because bioinformatic analysis suggested a deleterious effect. p.Met451Val was considered of unknown significance because it was classified as benign by pathogenicity prediction software and, although it was absent from the control populations analyzed, the amino acid affected is not evolutionarily well conserved. These findings seemed to indicate that this variant is probably benign. None of the patients met the criteria for small patella syndrome (Table 3 and of the ).
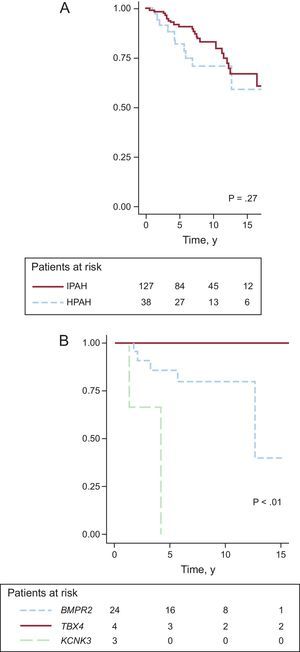
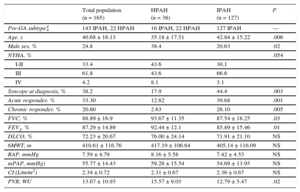
Prognostic Impact of the Genetic Alterations (Kaplan-Meier Survival Analysis)The mean follow-up was 7.74 ± 4.46 years. Survival analysis failed to show significant differences between IPAH and HPAH (Figure A) and only showed differences among the HPAH forms associated with TBX4, KCNK3, and BMPR2 (Figure B).
Analysis of the Factors Predicting Death or Transplant (Cox Regression)
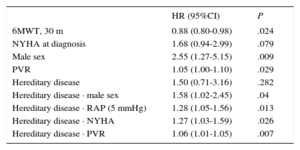
The results of the univariate and multivariate analyses are shown in Table 4 and Table 5 (log likelihood function = –103.29; log-rank chi-square = 13.03; P = .005).
Cox Univariate Analysis
| HR (95%CI) | P | |
|---|---|---|
| 6MWT, 30 m | 0.88 (0.80-0.98) | .024 |
| NYHA at diagnosis | 1.68 (0.94-2.99) | .079 |
| Male sex | 2.55 (1.27-5.15) | .009 |
| PVR | 1.05 (1.00-1.10) | .029 |
| Hereditary disease | 1.50 (0.71-3.16) | .282 |
| Hereditary disease · male sex | 1.58 (1.02-2.45) | .04 |
| Hereditary disease · RAP (5 mmHg) | 1.28 (1.05-1.56) | .013 |
| Hereditary disease · NYHA | 1.27 (1.03-1.59) | .026 |
| Hereditary disease · PVR | 1.06 (1.01-1.05) | .007 |
6MWT, 6-minute walk test; 95%CI, 95% confidence interval; HR, hazard ratio; NYHA, New York Heart Association; PVR, pulmonary vascular resistance; RAP, right atrial pressure.
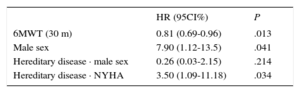
Cox Multivariate Analysis
| HR (95CI%) | P | |
|---|---|---|
| 6MWT (30 m) | 0.81 (0.69-0.96) | .013 |
| Male sex | 7.90 (1.12-13.5) | .041 |
| Hereditary disease · male sex | 0.26 (0.03-2.15) | .214 |
| Hereditary disease · NYHA | 3.50 (1.09-11.18) | .034 |
6MWT, 6-minute walk test; 95%CI, 95% confidence interval; HR: hazard ratio; NYHA, New York Heart Association.
The genetic study was refused by 2 of 19 families and accepted by 72 of the 90 family members (80%) belonging to the remaining 17 families. The study identified 21 healthy carriers (29.1%) and 6 affected carriers (8.3%). The characteristics of the carriers are shown in and of the . The estimated penetrance was 23.5% for BMPR2 mutations, 25.0% for KCNK3 mutations, and 33.0% for TBX4 mutations.
DISCUSSIONWith 165 patients, our series is the largest PAH patient series to be genetically studied in Spain, collecting 33% of idiopathic and hereditary forms registered with REHAP until November 20141 and 49% of patients treated in 3 referral centers. Mutations in BMPR2 were found in 50% of the hereditary forms and in 9% of the idiopathic forms of PAH. This rate is lower than the previously reported 75% and 25% for HPAH and IPAH respectively.2,4,5,7,12 Inclusion of the TBX4 and KCNK3 mutations increased the percentage of mutation carriers to 68.1% and 11.1%, respectively, approaching the data from other groups.12 This could indicate the presence of a different genetic background in the Spanish population, because the total percentage of patients with mutations was similar to that of other populations but showed a distinct distribution of the affected genes.4,7,12,19,22 However, more extensive studies are required to confirm this hypothesis.
Our work describes the phenotype associated with each type of BMPR2 alteration, as well as the lesser known forms associated with the KCNK3 and TBX4 genes. Analysis of our data revealed a more aggressive profile of the forms associated with KCNK3 vs the more benign behavior of TBX4, with an intermediate clinical profile for BMPR2. However, similar to previous reports,12 there was no difference in survival between the hereditary and idiopathic forms. One of the most important findings of this study is the marked prognostic impact of the hereditary forms on the diagnosis of an advanced functional class, highlighting the particular importance of an early diagnosis in this subgroup of patients.
Hereditary Pulmonary Arterial Hypertension vs Idiopathic Pulmonary Arterial HypertensionIn our series, the clinical profile of hereditary forms as a whole was similar to that associated with BMPR2.4,7,9,12,19 Notably, in 31.81% of patients with HPAH, the genetic study failed to identify the causative mutation; this percentage is higher than in previous reports.2,3
Mutations in BMPR2Of the 19 mutations identified in BMPR2, 42% were missense mutations, a higher rate than previously reported.4 Of these, 11 were novel, showing the complexity of this gene, which continues to show new alterations5,20 despite having been widely studied. Although these changes were distributed throughout the gene and affected various protein domains, 8 of them (42%) were located in exon 2, which was a much higher frequency than previously described.4 This finding could indicate the presence of a mutation “hot spot” in this exon, at least in the Spanish population.
The identification of the c.653_676dupATAAAGGCTCCTTGGATGAGCG mutation, a duplication of 22 base pairs, is notable because this type of change has not previously been linked to PAH development. This mutation was characterized by a higher penetrance (50.0% vs 23.5%), a novel finding with possible future implications.
The previous findings on the phenotype associated with BMPR2 were confirmed in our series (Table 2),4–9,20 except that there was a lower proportion of women with the hereditary form of the disease, another novel finding. The “respiratory advantage” of the forms associated with BMPR2 described by Girerd23,24 was confirmed, absent from the idiopathic forms and the other hereditary forms. This finding could reflect a pathophysiological process distinct at the pulmonary vascular level related to the BMPR2/Smad pathway. There were no significant differences in the phenotype according to the type of BMPR2 mutation, in contrast to the findings of other studies.8
Mutations in KCNK3So far, 9 cases of PAH have been linked to KCNK3: 6 in the series of Ma et al.13 and 3 in our series, with 2 novel pathogenic variants. The identification of further mutations in KCNK3 is expected, given that the gene has only relatively recently been linked to PAH. Our group has described for the first time a homozygous mutation in KCNK3 (p.Gly106Arg) in a patient with consanguineous parents and an aggressive form of the disease; the mother is a heterozygous carrier of this mutation. Although further confirmatory studies are required, the presence of both mutated alleles in the index case could explain the aggressive symptoms.
Similar to the work of Ma et al.,13 our study suggests the presence of a more aggressive phenotype of the forms associated with KCNK3, even if they share certain characteristics with the forms associated with BMPR2, such as autosomal dominant inheritance, incomplete penetrance, and predominantly adult onset. Various experimental approaches are being attempted to reestablish the functioning of this channel,25,26 possibly opening a new line of treatment for these patients.
Mutations in TBX4Besides our current series with 4 patients with pathogenic mutations in TBX4, the only other known patients with TBX4 mutations are the 7 patients reported by Kerstjens-Frederikse et al.14 Notably, all of our patients with childhood-onset PAH showed changes in exon 8, whereas the patients with adult-onset PAH had mutations in exon 3. This phenomenon was not seen in the series of Kerstjens-Frederikse et al.,14 whose patients with childhood-onset PAH showed variations in either of the 2 exons.
The phenotype of this subgroup of patients has been poorly described, although it has been suggested that this form shows predominantly childhood onset and lower aggressiveness and is often associated with small patella syndrome. However, none of our patients met the diagnostic criteria for this skeletal dysplasia. Our results support these findings, with no death or transplant event after a mean follow-up of 21.75 years, despite the baseline hemodynamic severity. Thus, PAH associated with mutations in TBX4 could represent the most benign form of this disease, although further studies are required to confirm this hypothesis.
Analysis of Prognostic FactorsIn the population with IPAH, univariate analysis showed a prognostic impact of the classical prognostic factors derived from population registries, such as male sex, 6-minute walking distance, and arterial pulmonary vascular resistance. These results are similar to those of previous reports,27,28 even if the impact of hereditary disease and the baseline NYHA functional class were not significant, in contrast to previous findings (Table 4)12,27,28, of the . Only the 6-minute walking distance and male sex were still significant in the multivariate analysis. In HPAH patients, male sex and the classical prognostic parameters, related to advanced stages of the disease (right atrial pressure, elevated vascular resistance, and advanced functional class), also had a negative impact in the univariate analysis, although only the functional class was still significant in the multivariate analysis of this subgroup. Thus, NYHA functional class III or IV in the HPAH subgroup was related to a 3.5 times higher risk of placement on the lung transplant list or death (Table 5), a novel finding indicating the importance of an early diagnosis.
Familial ImpactWe obtained a significant percentage of negative results in the genetic and echocardiographic screening of the family members, as published by other groups.28 However, due to the prognostic impact of early diagnosis, particularly in the hereditary forms, appropriate screening of family members is vital to detect asymptomatic carriers at an early stage. Thus, of particular interest is the development of follow-up programs of healthy carriers, a field being intensively investigated.28–30
Regarding the penetrance of the alterations described, incomplete penetrance with considerable interfamily variability was observed in the 3 genes studied in our population. This finding highlights the marked differences according to the type of mutation in the forms associated with BMPR2, even if the global penetrance of BMPR2 in our series is almost identical to that of previous findings.6,19 Our group is the first to describe the penetrance associated with mutations in TBX4 and further studies are required to confirm our results.
LimitationsAlthough the present study has analyzed the largest Spanish cohort of patients with PAH, our findings should be interpreted with caution, largely because both prevalent and incident cases were included, which might have caused a significant dispersion in the date of the diagnosis with a possible underlying survival bias. In addition, just 3 of the genes related to the development of the disease were studied, including BMPR2, the main gene linked to the disease, and the many other genes linked to the disease should be included in future studies.13,15,16 The categorization of the pathogenicity of the mutations found was an estimate based on diverse clinical and genetic/molecular parameters. Thus, for their confirmation, further work is required that demonstrates cosegregation of the mutation with the phenotype and/or involves functional studies.
CONCLUSIONSThe present study has identified a distinguishing feature in the Spanish population with PAH with respect to other described populations, with a lower percentage of BMPR2-associated forms and the presence of a possible novel hot spot in exon 2 of this gene. In addition, the findings from our population suggest that the forms associated with TBX4 might show a more benign presentation, in contrast to the more aggressive phenotype of the forms associated with KCNK3. No differences were seen in prognosis between the idiopathic and hereditary forms, and our study also showed the significance of the classical prognostic factors of PAH. Our results also highlight the considerable prognostic impact of early diagnosis in the hereditary forms of the disease with the consequent particular importance of adequate screening of this patient subgroup. New studies are required to confirm these findings in order to incorporate the phenotypic and prognostic nuances of each genetic alteration into daily clinical practice.
FundingThis project was partially funded by the Cardiovascular Investigation Network of the Instituto de Salud Carlos III of Madrid (RD06/0003/0012), as well as by unconditional grants from the Spanish Pulmonary Arterial Hypertension Association, Actelion, and Foundation Air Liquide.
Conflicts of interestNone declared.
- –
For years, BMPR2 has been the main gene associated with PAH development, present in 75% of hereditary forms and 25% of idiopathic forms. Advances in genetics, such as massively parallel sequencing, have enabled the discovery of numerous other genes related to this disease, such as KCNK3 and TBX4. According to the published data, the forms associated with BMPR2 can be characterized by a younger age at diagnosis, greater hemodynamic severity, and reduced response to vasodilator testing, without evidence of prognostic differences. However, the phenotype and prognosis associated with TBX4 and KCNK3 have been scarcely described.
- –
This work involves the largest series of patients with idiopathic and hereditary PAH genetically studied in Spain, as well as the largest study of family members published in Spain in this field. We analyzed the prevalence and genotype-phenotype correlations of mutations in BMPR2, TBX4, and KCNK3 in Spain. This is the first study to describe PAH caused by a duplication in BMPR2 and the first patients with a TBX4 mutation without associated bone disease. Finally, we observed a marked prognostic impact of late diagnosis in the hereditary forms, an important finding underscoring the value of early diagnosis in this patient subgroup.
We would like to thank the Spanish Pulmonary Arterial Hypertension Association, Actelion, Air Liquide Foundation, the Biobanc tissue bank of the Hospital Clínic de Barcelona, and our patients for making this study possible. In addition, we thank the Exome Aggregation Consortium and the NHLBI (National Heart, Lung, and Blood Institute) GO Exome Sequencing Project and their ongoing projects (Lung GO Sequencing Project [HL-102923], WHI Sequencing Project [HL-102924], Broad GO Sequencing Project [HL-102925], Seattle GO Sequencing Project [HL-102926], and Heart GO Sequencing Project [HL-103010]) for their invaluable contributions to the scientific community.