Activation of both the sympathetic nervous system and the renin-angiotensin-aldosterone system is closely associated with pulmonary arterial hypertension. We hypothesized that renal denervation decreases renin-angiotensin-aldosterone activity and inhibits the progression of pulmonary arterial hypertension.
MethodsTwenty-two beagles were randomized into 3 groups. The dogs’ pulmonary dynamics were measured before and 8 weeks after injection of 0.1mL/kg dimethylformamide (control dogs) or 2mg/kg dehydromonocrotaline (pulmonary arterial hypertension and pulmonary arterial hypertension + renal denervation dogs). Eight weeks after injection, neurohormone levels and pulmonary tissue morphology were measured.
ResultsLevels of plasma angiotensin II and endothelin-1 were significantly increased after 8 weeks in the pulmonary arterial hypertension dogs and were higher in the lung tissues of these dogs than in those of the control and renal denervation dogs (mean [standard deviation] angiotensin II: 65 [9.8] vs 38 [6.7], 46 [8.1]; endothelin-1: 96 [10.3] vs 54 [6.2], 67 [9.4]; P < .01). Dehydromonocrotaline increased the mean pulmonary arterial pressure (16 [3.4] mmHg vs 33 [7.3] mmHg; P < .01), and renal denervation prevented this increase. Pulmonary smooth muscle cell proliferation was higher in the pulmonary arterial hypertension dogs than in the control and pulmonary arterial hypertension + renal denervation dogs.
ConclusionsRenal denervation attenuates pulmonary vascular remodeling and decreases pulmonary arterial pressure in experimental pulmonary arterial hypertension. The effect of renal denervation may contribute to decreased neurohormone levels.
Keywords
Pulmonary arterial hypertension (PAH) is a fatal disease characterized by excessive pulmonary vascular remodeling that leads to elevated pressure in the pulmonary vascular system and in the right side of the heart.1 Although the pathogenesis of PAH remains incompletely understood, previous studies have implicated the increased activity of the sympathetic nerve system and the renin–angiotensin–aldosterone system.2,3 It has been suggested that increased sympathetic nerve activation could contribute to the alveolar hyperventilation observed in patients with PAH.4,5 The increased plasma levels of renin and angiotensin II (Ang II) have been closely associated with PAH progression and prognosis.6 Numerous treatments have been proven to be useful in decreasing PAH, but no treatment has long-term effects.7,8
Previous studies have confirmed that a significant reduction in renal noradrenaline spillover and blood pressure can be achieved after catheter-based renal sympathetic denervation (RSD).9,10 Our previous study demonstrated that plasma Ang II concentrations were attenuated after RSD.11,12 It is unknown whether RSD has an impact on the other neurohormones and the progression of PAH. Therefore, in the present study, we examined the impact of RSD on pulmonary vascular remodeling and neurohormones in experimental PAH.
METHODSPreparation of the Animal ModelTwenty-two beagles of either sex, with a mean (standard deviation) weight of 13.5 (2.4) kg, were used in the present study. This study conformed to the current Guide for the care and use of laboratory animals, published by the National Institutes of Health (no. 85-23, revised in 1996). The study protocol was approved by the Ethics Committee of Wuhan University. Animal handling was performed according to the Wuhan Directive for Animal Research.
An intramuscular injection of 25mg/kg ketamine sulfate was administered before pentobarbital sodium premedication. All the dogs were administered pentobarbital sodium (30mg/kg intravenously), intubated, and ventilated with room air supplemented with oxygen by a respirator (MAO01746, Harvard Apparatus; Holliston, Montana, United States). Continuous electrocardiogram monitoring was performed using leads I, II and III. Group 1 consisted of 7 dogs that received 0.1mL/kg dimethylformamide. Group 2 comprised 8 dogs that received dehydromonocrotaline (DHMC). Group 3 comprised 7 dogs that received DHMC and renal artery ablation. Group 1 was assigned to the control group (to exclude the effect of dimethylformamide on PAH, we used dimethylformamide as control), group 2 to the PAH group, and group 3 to the PAH+RSD group.
Dehydromonocrotaline was artificially synthesized as described by Mattocks et al.13 The purity of the DHMC was determined by high-performance liquid chromatography.14 Dehydromonocrotaline was dissolved in 0.1mL/kg dimethylformamide just before injection.
Study ProtocolAfter stable anesthesia was obtained, all the beagles were injected with 1000 U heparin and hemostatic sheaths were inserted into their right femoral veins. Under X-ray fluoroscopy guidance, a 6-Fr Swan-Ganz pulmonary artery catheter (Edwards Lifesciences; Irvine, California, United States) filled with heparinized saline was inserted through the vein. The catheter was connected to both a pressure transducer and a Vigilance monitoring system. The pulmonary artery catheter was delivered through the right atria and ventricle into the small pulmonary artery. Then, the catheter was withdrawn, and measurements were taken of the pulmonary capillary wedge pressure, pulmonary arterial systolic pressure, pulmonary artery mean pressure, right ventricular systolic pressure, and right ventricular mean pressure. Cardiac output was measured using the continuous thermodilution method with the Vigilance monitoring system. We calculated the pulmonary vascular resistance according to the formula (pulmonary vascular resistance = 80 × [pulmonary artery mean pressure – pulmonary capillary wedge pressure]/cardiac output). After the baseline hemodynamic measurements, PAH and PAH+RSD beagles were injected with 2mg/kg DHMC, and the control beagles were injected with 0.1mL/kg dimethylformamide via a Swan-Ganz pulmonary artery catheter inserted into the right atria. In the PAH+RSD group, after the baseline hemodynamic measurements, the procedure of RSD was performed as in a previous study.15 Then, all the beagles were allowed to recover for 8 weeks. After 8 weeks, all hemodynamic measurements were repeated in the 3 groups.
EchocardiographyTransthoracic 2-dimensional and Doppler echocardiography were performed in all the animals (IE33, S5-1, PHILIPS; The Netherlands) at baseline and again after 8 weeks. Standard 2-dimensional short-and long-parasternal views and 4-, 2-, and 3-chamber apical views were obtained. The left atrial dimension, right atrial dimension, left ventricular diastolic dimension, and right ventricular diastolic dimension were measured using Simpson's biplane formula. All volumes were measured in triplicate, and the averages were reported. The right ventricular end-systolic longitudinal strain was analyzed by 2-dimensional speckle tracking. An independent echocardiography expert reviewed the images and the parameters.
Biochemical StudiesFour milliliters of venous blood was collected in EDTA (ethylenediaminetetraacetic acid) Vacutainer tubes and centrifuged at 2310g for 10minutes at 4°C (Beckman Coulter, Avanti J-E) at baseline and again after 8 weeks. The serum was separated, kept in microtubes, and stored at −80°C until assay. Levels of creatinine, Ang II, prostaglandin E2, and endothelin-1 (ET-1) were examined by ELISA. The inferior lobe of the left lung was isolated after thoracotomy. Levels of Ang II, prostaglandin E2 and ET-1 were measured in the lung as previously described.16 Tissues were obtained from the right ventricular base in all animals after 8 weeks. Levels of aldosterone and B-type natriuretic peptide were measured by ELISA.
Histological StudiesThe excised left lungs were processed for light microscopic observation according to standard procedures. Tissue samples were chopped into small pieces, embedded in paraffin, cut into 3-μm slices and stained with hematoxylin and eosin. The sections were observed, analyzed, and photographed using a Nikon eclipseCi light microscope. The pulmonary arteries were identified as vessels with 2 clearly defined elastic laminae, with a layer of smooth muscle cells between the 2 laminae. The wall thickness of the arteries was measured for 15 muscular arteries at 400 magnification.
Expression of Ang II type 1 (AT1) receptor and Ang II type 2 (AT2) receptors in the pulmonary artery was measured using Western blot. The membranes were blocked with 5% nonfat dry milk in Tris-buffered saline with Tween for 1h and incubated overnight at 4°C with the primary antibodies (monoclonal rabbit anti-AT1 and anti-AT2 antibodies [Santa Cruz Biotechnology Inc.; Dallas, Texas, United States]; used at 1:500; rabbit antiactin antibody, antibody [Santa Cruz Biotechnology Inc]; used at 1:1000). They were then washed in tris buffered saline tween 3 times, incubated with second antibody for 1h at 37°C, and revealed by Immun-Star horseradish peroxidase substrate. The relative expression of protein was determined with image analyzer software (AlphaEase FC, United States).
Sections of ventricle were dissected from the heart and rapidly stored at −80oC. Right ventricular sections were taken from the right ventricular outflow. Masson's trichrome staining was used to identify increased concentration of interstitial fibrosis. Connective tissue was differentiated on the basis of its color and expressed as a percentage of the reference tissue area (NIKON Ti-s, Japan). Blood vessels and perivascular interstitial cells were excluded from the connective tissue quantification. Interstitial collagen volume fraction in the ventricular was determined by quantitative morphometry with an image analyzer (IPP 6.0, Media Cybemetics; Georgia, United States).
Statistical AnalysisValues are shown as means (standard deviation). The echocardiography results of the PAH and PAH+RSD groups were compared for the poststudy period using the paired t test, and 2-sample independent Student's t tests were used to compare the means of the 2 groups. Analysis of variance in the form of Neuman-Keuls tests were used to compare the means of the continuous variables among multiple groups, and any significant difference was further analyzed using the Tukey–Kramer test. All statistical tests were 2-sided, and a probability value < .05 was required for statistical significance.
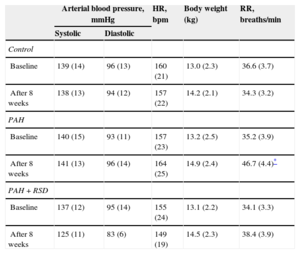
RESULTSCharacteristics of Animal ModelsThere were no significant differences in arterial blood pressure, heart rate or body weight at baseline among the 3 experimental groups. In the PAH+RSD group, systolic blood pressure was decreased after 8 weeks, but this decrease was not statistically significant. The PAH dogs began to display rapid breathing and decreased appetite beginning at 10 days after injection. Lung color was paler in the PAH and PAH+RSD dogs than in the control dogs. The characteristics at baseline and after 8 weeks are shown in Table 1.
Characteristics of Animal Models
| Arterial blood pressure, mmHg | HR, bpm | Body weight (kg) | RR, breaths/min | ||
|---|---|---|---|---|---|
| Systolic | Diastolic | ||||
| Control | |||||
| Baseline | 139 (14) | 96 (13) | 160 (21) | 13.0 (2.3) | 36.6 (3.7) |
| After 8 weeks | 138 (13) | 94 (12) | 157 (22) | 14.2 (2.1) | 34.3 (3.2) |
| PAH | |||||
| Baseline | 140 (15) | 93 (11) | 157 (23) | 13.2 (2.5) | 35.2 (3.9) |
| After 8 weeks | 141 (13) | 96 (14) | 164 (25) | 14.9 (2.4) | 46.7 (4.4)* |
| PAH+RSD | |||||
| Baseline | 137 (12) | 95 (14) | 155 (24) | 13.1 (2.2) | 34.1 (3.3) |
| After 8 weeks | 125 (11) | 83 (6) | 149 (19) | 14.5 (2.3) | 38.4 (3.9) |
bpm, beats per minute; HR, heart rate; PAH, pulmonary arterial hypertension; RR, respiratory rate; RSD, renal sympathetic denervation.
Data are expressed as mean (standard deviation).
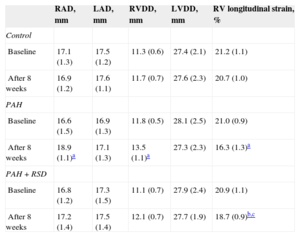
In contrast, right atrial dimension significantly increased after 8 weeks in the PAH dogs (P = .02). No significant differences in the right atrial dimension were observed prior to or after 8 weeks in the control and PAH+RSD groups. Significant increases in the right ventricular diastolic dimension were also noted in the PAH dogs. The right ventricular diastolic dimension was increased in PAH+RSD group after 8 weeks, but this increase was not statistically significant (P = .09). After 8 weeks, the longitudinal strain of right ventricular lateral wall was reduced in PAH group and PAH+RSD group. However, the longitudinal strain was higher in the PAH+RSD group than in the PAH group (Table 2).
Changes of Echocardiographic Parameters at Baseline and After 8 Weeks in the 3 Groups
| RAD, mm | LAD, mm | RVDD, mm | LVDD, mm | RV longitudinal strain, % | |
|---|---|---|---|---|---|
| Control | |||||
| Baseline | 17.1 (1.3) | 17.5 (1.2) | 11.3 (0.6) | 27.4 (2.1) | 21.2 (1.1) |
| After 8 weeks | 16.9 (1.2) | 17.6 (1.1) | 11.7 (0.7) | 27.6 (2.3) | 20.7 (1.0) |
| PAH | |||||
| Baseline | 16.6 (1.5) | 16.9 (1.3) | 11.8 (0.5) | 28.1 (2.5) | 21.0 (0.9) |
| After 8 weeks | 18.9 (1.1)a | 17.1 (1.3) | 13.5 (1.1)a | 27.3 (2.3) | 16.3 (1.3)a |
| PAH+RSD | |||||
| Baseline | 16.8 (1.2) | 17.3 (1.5) | 11.1 (0.7) | 27.9 (2.4) | 20.9 (1.1) |
| After 8 weeks | 17.2 (1.4) | 17.5 (1.4) | 12.1 (0.7) | 27.7 (1.9) | 18.7 (0.9)b,c |
LAD, left atrial dimension; LVDD, left ventricular diastolic dimension; PAH, pulmonary arterial hypertension; RAD, right atrial dimension; RSD, renal sympathetic denervation; RV, right ventricular; RVDD, right ventricular diastolic dimension.
Data are expressed as mean (standard deviation).
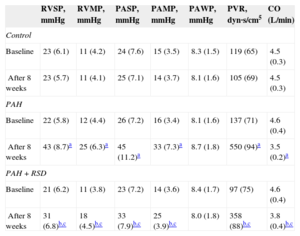
The hemodynamic data at baseline and after 8 weeks among the 3 groups are shown in Table 3. There was no significant difference in the baseline pulmonary hemodynamic indices among the 3 groups. Compared with baseline, pulmonary artery pressure and right ventricular pressure were higher after 8 weeks in the PAH group. There was no significant difference in the hemodynamic parameters at baseline and after 8 weeks in the control group. The pulmonary artery pressure and right ventricular pressure increased after 8 weeks in the PAH+RSD group, but these hemodynamic parameters were lower in the PAH+RSD dogs than in PAH dogs.
Changes of Hemodynamic Parameters at Baseline and After 8 Weeks in the 3 Groups
| RVSP, mmHg | RVMP, mmHg | PASP, mmHg | PAMP, mmHg | PAWP, mmHg | PVR, dyn·s/cm5 | CO (L/min) | |
|---|---|---|---|---|---|---|---|
| Control | |||||||
| Baseline | 23 (6.1) | 11 (4.2) | 24 (7.6) | 15 (3.5) | 8.3 (1.5) | 119 (65) | 4.5 (0.3) |
| After 8 weeks | 23 (5.7) | 11 (4.1) | 25 (7.1) | 14 (3.7) | 8.1 (1.6) | 105 (69) | 4.5 (0.3) |
| PAH | |||||||
| Baseline | 22 (5.8) | 12 (4.4) | 26 (7.2) | 16 (3.4) | 8.1 (1.6) | 137 (71) | 4.6 (0.4) |
| After 8 weeks | 43 (8.7)a | 25 (6.3)a | 45 (11.2)a | 33 (7.3)a | 8.7 (1.8) | 550 (94)a | 3.5 (0.2)a |
| PAH+RSD | |||||||
| Baseline | 21 (6.2) | 11 (3.8) | 23 (7.2) | 14 (3.6) | 8.4 (1.7) | 97 (75) | 4.6 (0.4) |
| After 8 weeks | 31 (6.8)b,c | 18 (4.5)b,c | 33 (7.9)b,c | 25 (3.9)b,c | 8.0 (1.8) | 358 (88)b,c | 3.8 (0.4)b,c |
CO, cardiac output; PAH, pulmonary arterial hypertension; PAMP, pulmonary artery mean pressure; PASP, pulmonary arterial systolic pressure; PAWP, pulmonary capillary wedge pressure; PVR, pulmonary vascular resistance; RSD, renal sympathetic denervation; RVMP, right ventricular mean pressure; RVSP, right ventricular systolic pressure.
Data are expressed as mean (standard deviation).
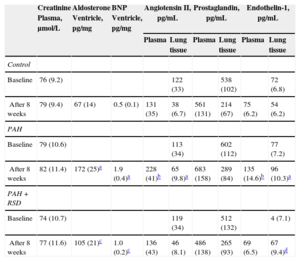
The PAH group showed a statistically significant increase in the plasma and lung tissue levels of Ang II at the endpoint of the protocol (P < .01) compared with baseline (Table 4). There were no significant differences in the plasma levels of Ang II between the baseline and the study endpoint in the control and PAH+RSD groups. There were no significant differences in the plasma creatinine and lung tissue levels of prostaglandin E2 between baseline and the study endpoint in the 3 groups.
Changes of Neurohormone Concentrations in Blood, Lung and Ventricular Tissues at Baseline and After 8 Weeks in the 3 Groups
| Creatinine Plasma, μmol/L | Aldosterone Ventricle, pg/mg | BNP Ventricle, pg/mg | Angiotensin II, pg/mL | Prostaglandin, pg/mL | Endothelin-1, pg/mL | ||||
|---|---|---|---|---|---|---|---|---|---|
| Plasma | Lung tissue | Plasma | Lung tissue | Plasma | Lung tissue | ||||
| Control | |||||||||
| Baseline | 76 (9.2) | 122 (33) | 538 (102) | 72 (6.8) | |||||
| After 8 weeks | 79 (9.4) | 67 (14) | 0.5 (0.1) | 131 (35) | 38 (6.7) | 561 (131) | 214 (67) | 75 (6.2) | 54 (6.2) |
| PAH | |||||||||
| Baseline | 79 (10.6) | 113 (34) | 602 (112) | 77 (7.2) | |||||
| After 8 weeks | 82 (11.4) | 172 (25)a | 1.9 (0.4)a | 228 (41)b | 65 (9.8)a | 683 (158) | 289 (84) | 135 (14.6)b | 96 (10.3)a |
| PAH+RSD | |||||||||
| Baseline | 74 (10.7) | 119 (34) | 512 (132) | 4 (7.1) | |||||
| After 8 weeks | 77 (11.6) | 105 (21)c | 1.0 (0.2)c | 136 (43) | 46 (8.1) | 486 (138) | 265 (93) | 69 (6.5) | 67 (9.4)d |
BNP, B-type natriuretic peptide; PAH, pulmonary arterial hypertension; RSD, renal sympathetic denervation; SD, standard deviation.
Data are expressed as mean (standard deviation).
There was no significant difference at the baseline levels of serum ET-1 concentration among the 3 groups. After 8 weeks, serum ET-1 levels increased (P < .01) in the PAH dogs, but no statistically significant difference was observed in the control dogs. In the PAH+RSD group, the serum ET-1 concentration was decreased after 8 weeks, but this decrease was not statistically significant (P = .19). The ET-1 concentration in the lung tissue was significantly higher in the PAH group than in the control group (P < .01). Furthermore, the ET-1 concentration in the lung tissue was higher in the PAH+RSD group than in the control group (P = .01).
We further investigated levels of aldosterone and B-type natriuretic peptide in the right ventricle. The results showed that the levels of aldosterone and B-type natriuretic peptide in the right ventricular base tissue samples were higher in both groups with PAH induction than in control dogs. Interestingly, the levels of aldosterone and B-type natriuretic peptide in the right ventricle in the PAH group were higher than those in the PAH+RSD groups. This confirms that the PAH+RSD group had less severe right ventricle remodeling than the PAH group (Table 4).
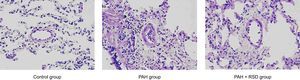
Morphological and Western-blot StudiesHematoxylin and eosin staining staining demonstrated significant intimal thickening and luminal stenosis in the PAH group compared with the control and PAH+RSD groups. Figure 1 shows the representative changes in the vascular structure for the 3 groups. The neointimal occlusion was evaluated using the vascular occlusion score. An average vascular occlusion score of the 30 selected vessels was calculated for each dog as an index of vascular occlusion. The results showed that neointimal lesions occurred in 64% of the chosen small pulmonary arteries in the PAH group. The mean vascular occlusion score was 1.17 in these arteries. In contrast, the dogs in the PAH+RSD group developed neointimal lesions in 27% of the selected pulmonary arteries, and the mean vascular occlusion score was 0.24. The difference between the PAH group and the PAH+RSD group was significant (1.17 [0.13] vs 0.24 [0.10]; P < .01). The small pulmonary arteries in the control group showed that the mean vascular occlusion score was 0.
.")
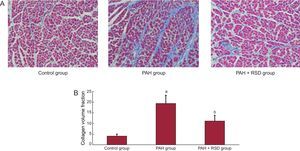
Figure 2 illustrates an example of Masson's trichrome staining in the tissue sections. In contrast, the ventricular sections from PAH+RSD animals had significantly less fibrosis than PAH dogs, while PAH+RSD animals had higher fibrosis than control dogs. For instance, images from PAH hearts revealed a large amount of fibrosis (19.4 [3.8%]), whereas control dogs showed minimal fibrous tissue (4.1 [0.9%]) and PAH+RSD dogs showed moderate fibrous tissue (11.2 [2.6%]).
. A: examples of the right ventricular outflow in the 3 groups; B: summary of changes in the right ventricular outflow. In contrast, the right ventricular sections in pulmonary arterial hypertension dogs had higher fibrosis than in control and pulmonary arterial hypertension+renal sympathetic denervation dogs, and the fibrosis in pulmonary arterial hypertension+renal sympathetic denervation dogs was higher than that in control dogs. PAH, pulmonary arterial hypertension; RSD, renal sympathetic denervation. aP<.01 vs control group and pulmonary arterial hypertension+renal sympathetic denervation group. bP<.01 vs control group.")
Representative of histological changes in the 3 groups. Red areas represent myocytes and blue areas represent collagen (×400). A: examples of the right ventricular outflow in the 3 groups; B: summary of changes in the right ventricular outflow. In contrast, the right ventricular sections in pulmonary arterial hypertension dogs had higher fibrosis than in control and pulmonary arterial hypertension+renal sympathetic denervation dogs, and the fibrosis in pulmonary arterial hypertension+renal sympathetic denervation dogs was higher than that in control dogs. PAH, pulmonary arterial hypertension; RSD, renal sympathetic denervation. aP<.01 vs control group and pulmonary arterial hypertension+renal sympathetic denervation group. bP<.01 vs control group.
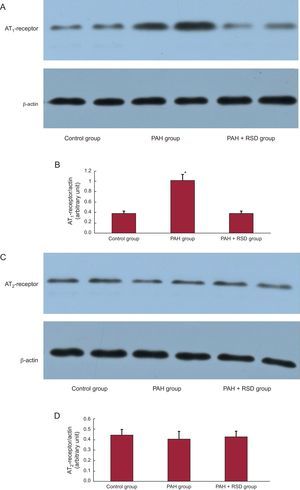
Figure 3 compares the Western blots of the pulmonary artery in the 3 groups. All immunoblot band intensity measurements were normalized to the β-actin band intensity of the loaded sample. Band densities were determined and allowed for the relative quantification of AT1 and AT2 densities. As shown in Figures 3A and B, the expression of AT1 in the pulmonary artery was higher in the PAH group than in the control and PAH+RSD groups (1.02 [0.11] vs 0.39 [0.04] and 0.38 [0.05]; P < .01). There were no significant difference in AT2 densities in the 3 groups (0.44 [0.04] vs 0.41 [0.05] and 0.42 [0.04]; P = .38) (Figures 3C and D).

Western blot analysis of angiotensin II type 1-receptor and angiotensin II type 2-receptor in the protein samples extracts from the pulmonary artery. A: examples of the Western blot bands recognized by the angiotensin II type 1-receptor antibodies. B: mean band densities expressed as the ratios of the 3 groups. The pulmonary arterial hypertension group over the control group and the pulmonary arterial hypertension+renal sympathetic denervation group. C: examples of the Western blot bands recognized by angiotensin II type 2-receptor antibodies. D: mean band densities expressed as the ratios of the 3 groups. There was no significant difference in angiotensin II type 2 densities in the 3 groups. AT1, angiotensin II type 1; AT2, angiotensin II type 2; PAH, pulmonary arterial hypertension; RSD, renal sympathetic denervation. *P<.01.
The present study explored the impact of RSD on pulmonary vascular remodeling in experimental PAH. We provide evidence: a) that RSD attenuated the PAH induced by DHMC treatment in the beagles, as evidenced by the absence of a significant increase in pulmonary artery pressure, vascular wall thickness, and increased cardiac output, and b) that decreased levels of Ang II and ET-1 after RSD were associated with pulmonary vascular remodeling, which was induced by the DHMC treatment. The present findings suggest that catheter-based RSD is a potentially effective alternative for PAH treatment.
Pulmonary arterial hypertension is characterized by elevations in pulmonary artery pressure and pulmonary vascular resistance. Multiple factors, including vasoconstriction, pulmonary vessel remodeling, and thrombosis, are involved in the pathogenesis of PAH.17,18 Sympathetic nerve activity is also an important causal factor in the development of PAH.2,19 In the animal model of acute PAH, the increased pulmonary artery pressure were completely abolished by pulmonary artery denervation.20,21 In patients with PAH, it has been suggested that systemic renin–angiotensin–aldosterone system activity is upregulated, based on findings of upregulated SNS activity. Previous studies have shown that Ang II can cause the growth/proliferation and hypertrophy of pulmonary artery muscle cells, thereby inducing pulmonary arterial resistance.22,23
In our previous studies, we conducted tests to measure Ang II levels before and after RSD. We found that the decreased release of Ang II demonstrated the effectiveness of the device in achieving an efferent RSD.12,24 To investigate whether RSD has an impact on pulmonary vascular remodeling, the experimental PAH model was investigated in the present study. Monocrotaline is a pyrrolizidine alkaloid plant toxin that, when activated, causes structural remodeling of pulmonary blood vessels and an increase in pulmonary arterial pressure.25,26 Our results are in accordance with previous investigations showing that the pulmonary artery pressure and right ventricular pressure in PAH dogs were significantly higher and that the cardiac output was lower than in the control dogs. Interestingly, in the present study, we found that the pulmonary artery pressure and right ventricular pressure in the PAH+RSD dogs were significantly lower than those in the PAH dogs. Furthermore, the pulmonary arterial wall thickening was decreased after RSD. A previous study has shown that plasma levels of circulating ET-1 are elevated in animals treated with monocrotaline.27 Our data shows that RSD decreases plasma Ang II and ET-1 levels in lung tissue. The circulating plasma levels of ET-1 are elevated in patients with PAH, and increased circulating levels of ET-1 correlate with increased pulmonary vascular resistance and increased mortality in patients with PAH.23,28 Early studies by Nakamura and Johns29 reported that a mild stimulation of the renal nerve increased angiotensinogen levels in the rat kidney. In another study, Girchev et al30 demonstrated that RSD decreases the papillary ET-1 concentration in Wistar-Kyoto rats. These results showed that renal nerves participate in the regulation of ET-1 production within the kidney.30 A recent study demonstrated that RSD prevented aortic regurgitation-induced increases in kidney norepinephrine and Ang II levels.31 Angiotensin II infusion led to an enhanced vascular ET that converts enzyme activity and renal ET-1 content in the rat. These results suggest that Ang II is a strong stimulus of ET-1 synthesis.32
Interestingly, in the present study, we found that AT1-receptor expression was upregulated in experimental PAH, but this change was suppressed after RSD. The effect of Ang II on PAH is mediated via AT1 receptors. De Man et al6 have demonstrated that enhanced AT1-receptor signaling was associated with increased pulmonary artery smooth muscle cell proliferation in PAH patients compared with controls. These data suggest the concept that, after RSD, the activity of the systemic renin–angiotensin–aldosterone system was inhibited, leading to local renin–angiotensin–aldosterone system-activity and AT1-receptor expression downregulation. Taken together, our results suggested that RSD treatment, prior to induction of PAH, prevented increases in right ventricular systolic pressure, and attenuated thickening of pulmonary vessels. The influence of RSD on PAH is mediated mainly by the activity of the renin–angiotensin–aldosterone and ET systems.
Potential SignificancePulmonary arterial hypertension is a rapidly progressive disease that ultimately leads to right-heart failure and death. Advances in understanding the pathobiology over the past 2 decades have led to therapies directed at reversing pulmonary vasoconstriction. Despite these advances, disease progression is common, even with the use of combination regimens targeting multiple mechanistic pathways. We showed that the pulmonary artery pressure was attenuated by RSD in experimental PAH. Renal sympathetic denervation may alter pulmonary vascular remodeling by inhibiting the renin–angiotensin–aldosterone system and ET activity. Our data is preliminary but renal artery ablation may be an effective alternative for the treatment of PAH patients. This finding might raise concerns about the conventional pathogenesis of PAH, and might suggest the effectiveness of RSD for PAH, a similar new understanding on the effect of pulmonary artery denervation for treatment of PAH patients.33
LimitationsThe present study has several limitations. First, the time course of the hemodynamic and structural derangements induced by DHMC was not studied. Thus, it is unknown whether hemodynamic and structural remodeling begins shortly after DHMC injection and whether these alterations are irreversible. Second, we performed renal denervation at the same time as pharmacological induction of PAH. Thus, the effects of RSD when the PAH is well established, as in the clinical scenario, cannot be ascertained. However, our results suggested that RSD were efficacious at initial stages of PAH. Future studies should investigate the effects of RSD on established PAH. Third, in control and PAH dogs, the radiofrequency ablation catheter was not positioned in the renal arteries. The control and PAH groups were not real sham groups. However, our results revealed RSD not only attenuated hemodynamic changes, but suppressed pulmonary vascular remodeling. Our findings set the stage for further comprehensive mechanistic study. Fourth, we did not investigate the anatomic distribution of periarterial sympathetic nerves around renal arteries by dual immunofluorescence staining using antibodies targeted for antityrosine hydroxylase and anticalcitonin gene–related peptide in control and RSD dogs. Whether all the plexus were destroyed around the renal artery is unknown. Finally, Aguero et al34 reported that right ventricular remodeling occurred at the structural, histological, and molecular level in a pulmonary vein banding model of chronic postcapillary pulmonary hypertension. In our study, we found that the right ventricle had higher levels of aldosterone, B-type natriuretic peptide, and interstitial fibrosis in PAH dogs. However, we did not investigate the changes of right ventricular mass and molecular level in experimental PAH by DHMC. The structural and molecular changes in the right ventricle in experimental PAH by DHMC should be further investigated.
CONCLUSIONSRenal sympathetic denervation suppressed pulmonary vascular remodeling and the decreased pulmonary arterial pressure in experimental PAH. The effect of RSD on PAH may contribute to the decreased levels of neurohormones.
FundingThis study was supported by the National Science and Technology Pillar Program of China (2011BAI11B12), the National Key Basic Research Development Program of China (The “973” Program, 2012CB518604) and the National Natural Science Foundation of China (81070144).
CONFLICTS OF INTERESTNone declared.