To the Editor:
Torsades de pointes (TdP) is a polymorphic ventricular tachycardia that appears with a long QT interval and can degenerate to ventricular fibrillation and sudden death. Long QT syndrome (LQT) is caused by alterations in cell membrane ionic channels, and it plays an important role in blocking the potassium channels codified by the hERG gene (IKr). It may be congenital, but it is more commonly acquired; medications, hydroelectrolyte alterations, and neurological, cardiac, and (more rarely) endocrine disorders are its most frequent causes.1,2
We present the case of a male patient 68 years of age who was referred to the emergency room for low level of consciousness and fever. His family reported 2 episodes of "absence" with disorientation, a fever spike of 38º and cold symptoms during the preceding days. His summarised personal history included atrial fibrillation (AF), arterial hypertension (treated with 50 mg losartan and 300 mg acetylsalicylic acid daily) and a transurethral resection 3 years before due to a papillary bladder growth (normal pre-op electrocardiogram).
The physical examination showed that the patient had no focal neurological signs, slowed thought process and coherent language; he was pale, with dry skin, arterial pressure of 72/41 mm Hg and a heart rate of 68 beats/min, no fever, and breathing well. Cardiopulmonary auscultation was normal, except for arrhythmic cardiac tones and an inflated abdomen with a homogeneous enlarged liver.
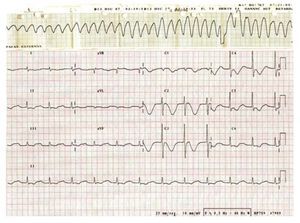
While he was being monitored, we witnessed TdP followed by VF, and thus proceeded to defibrillation and began a magnesium sulphate perfusion. Afterwards, the patient was admitted to the intensive care unit. The initial complementary tests showed an electrocardiogram in AF of 72 beats/ min with QTc of 705 ms (Figure 1) and laboratory analyses presenting the following: haemoglobin, 11.7 g/dL; leukocytes, 15 100/µL, 73% neutrophils; glucose, 96 mg/dL; urea, 61 mg/dL; creatinine, 2.4 mg/dL; sodium, 127 mmol/L; potassium, 4 mmol/L; total proteins, 7.2 g/dL; corrected calcium, 8.1 mg/ dL; magnesium, 1.6 mg/dL; GOT, 48 U/L; and GPT, 8 U/L. In addition, we ran a cranial computerised tomography (normal), lumbar puncture (clear fluid with red blood cells, leukocyte formula, and normal proteins), and an echocardiogram (left ventricle with a slight concentric hypertrophy with a good systolic function and biatrial dilation).

Figure 1. Image showing rhythm with torsades de pointes degenerating into ventricular fibrillation, and ECG after defibrillation in atrial fibrillation with a long QT interval (QTc, 705 ms).
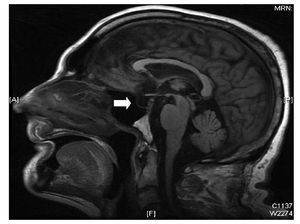
We began empirical antibiotic treatment with amoxicillin clavulanate, and ran an infectious disease and tumour screening (cephalospinal fluid culture, blood cultures, urine cultures, and tumour markers all negative; thoracic radiography, abdominal ultrasound, and proteinogram, all normal). The drugs were discontinued, renal function and hydroelectrolytic abnormalities were corrected, and we performed cardiac catheterisation (coronary arteries showed no significant lesions) and a cerebral MRI which revealed empty sella turcica, a pineal cyst 11 mm in diameter at its largest point, and a small area with a high T2 signal in the right mid cerebellar peduncle caused by a prior ischaemic event (8´3mm) (Figure 2). Given these findings, we called for a hormonal study, which showed panhypopituitarism: TSH, 0.239 (0.27-4.2) µU/mL; free T3, 0.95 (1.8-4.6) pg/mL; free T4, 0.79 (0.93-1.7) ng/dL; FSH, 0.81 (1.37-13.58) mUI/mL; LH, 0.16 (1.8-8.2) mUI/ml; total testosterone, <0.08 (1.6-8.7) ng/mL; 17-beta-estradiol, <10 (25-107) pg/mL; baseline cortisone, 1.26 (6.2-19.4) µg/dL. In light of these results, we began treatment with 100 µg/day levothyroxin and 10 mg/day hydrocortisone, which corrected the QT. Since a high risk of sudden death has been documented in patients with ventricular arrhythmias secondary to transitory or correctible causes,3 the case was presented in the clinical session and the decision was made to implant a defibrillator due to the risk of TdP recurrence under stressful circumstances. One month after discharge, the patient returned for a routine defibrillator checkup, and we witnessed AF with a QTc of 412 ms; he remained asymptomatic and had not presented arrhythmias.

Figure 2.Cerebral MRI showing empty silla turcica (arrow).
Empty sella turcica is a very rare cause of panhypopituitarism. The clinical profile tends to be insidious, and very few cases of TdP have been described.4 Because of their important hormonal interaction in the systemic and cardiovascular response to stress,2,5 the corticosteroid and thyroid deficits must be treated as early as possible.
We present the case of a patient with panhypopituitarism secondary to empty silla turcica, admitted with a clinical profile of acute suprarenal failure (hypovolaemic shock, hydroelectrolytic alterations, low level of consciousness, and fever)5 who developed TdP from LQT secondary to hormone deficit from the corticothyroid block and whose QT interval was corrected by hormone substitution treatment.