Nonischemic sudden cardiac death (SCD) is predominantly caused by cardiomyopathies and channelopathies. There are many diagnostic tests, including some complex techniques. Our aim was to analyze the diagnostic yield of a systematic diagnostic protocol in a specialized unit.
MethodsThe study included 56 families with at least 1 index case of SCD (resuscitated or not). Survivors were studied with electrocardiogram, advanced cardiac imaging, exercise testing, familial study, genetic testing and, in some cases, pharmacological testing. Families with deceased probands were studied using the postmortem findings, familial evaluation, and molecular autopsy with next-generation sequencing (NGS).
ResultsA positive diagnosis was obtained in 80.4% of the cases, with no differences between survivors and nonsurvivors (P=.53). Cardiac channelopathies were more prevalent among survivors than nonsurvivors (66.6% vs 40%, P=.03). Among the 30 deceased probands, the definitive diagnosis was given by autopsy in 7. A diagnosis of cardiomyopathy tended to be associated with a higher event rate in the family. Genetic testing with NGS was performed in 42 index cases, with a positive result in 28 (66.6%), with no differences between survivors and nonsurvivors (P=.21).
ConclusionsThere is a strong likelihood of reaching a diagnosis in SCD after a rigorous protocol, with a more prevalent diagnosis of channelopathy among survivors and a worse familial prognosis in cardiomyopathies. Genetic testing with NGS is useful and its value is increasing with respect to the Sanger method.
Keywords
Sudden cardiac death (SCD) is generally caused by a fatal ventricular arrhythmia such as ventricular fibrillation that progresses to death within a few minutes. Its most frequent etiology is ischemic heart disease but, in individuals younger than 35 years, the most 2 common causes–cardiomyopathy and channelopathy–have a genetic basis.1–3 This genetic etiology is increasingly more complex, with considerable heterogeneity and increasing numbers of causative genes.4,5 The usefulness of diagnostic protocols in determining the cause of SCD has been investigated in various studies. These protocols include both conventional techniques, such as electrocardiography (ECG) and echocardiography, and more complex techniques, such as cardiac magnetic resonance imaging, pharmacological testing, and genetic screening.6–12
Few studies have analyzed the diagnostic yields of the various tests according to survival from cardiac arrest. In addition, there is little information on the usefulness of genetic testing using next-generation sequencing (NGS) in selected SCD cases (molecular autopsy) and the familial penetrance of this genetic etiology.10–18 Our objective was to evaluate the diagnostic yield of a comprehensive protocol involving the stepwise use of conventional tests and more complex ones such as genetic screening using NGS in selected cases in an SCD population comprising both survivors and nonsurvivors of the cardiac arrest and to determine the penetrance of these diseases in the affected families.
METHODSNonischemic SCD cases were retrospectively analyzed in a specialized clinic. The sole inclusion criterion for survivors was the occurrence of a cardiac arrest requiring external defibrillation to restore sinus rhythm; in nonsurvivors, the death had to have occurred within 1 hour of symptom onset or less than 24hours after the individual was last seen alive, with no noncardiovascular cause of death. The exclusion criteria included an ischemic event, a secondary cause of the SCD, and patient or family refusal to participate in the registry. The SCD was considered ischemic based on the following criteria: in survivors, if there were indicators of acute coronary occlusion on ECG or coronary angiography or myocardial scarring in imaging tests; in nonsurvivors, if there was evidence of previous myocardial infarction with myocardial scarring or occlusive thrombi or the presence of atheromatous plaques in one or more coronary arteries with lumen occlusion > 75%. The study design was longitudinal and was approved by the local ethics committee.
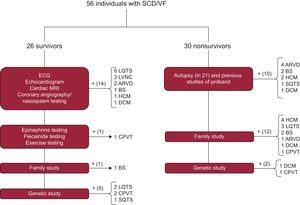
The diagnostic protocol is summarized in Figure 1.16 In the case of survivors, the battery of tests focused on the index case. Apart from use of medical history to identify precipitating factors, medication use, fever, and previous events, the first step comprised conventional studies such as resting and exercise ECG, transthoracic echocardiography, coronary angiography with and without vasospasm testing, and advanced cardiac imaging techniques. If no diagnosis could be made, the sequential diagnostic protocol described by our group in the FIVI-Gen study9 was applied to identify a concealed channelopathy. Briefly, pharmacological tests with epinephrine and flecainide were performed, followed by evaluation of first-degree family members using ECG or echocardiography; if all tests were negative, the index case was studied using genetic analysis.

Diagnostic algorithms for survivors and nonsurvivors of SCD. ARVD, arrhythmogenic right ventricular dysplasia; BS, Brugada syndrome; CPVT, catecholaminergic polymorphic ventricular tachycardia; DCM, dilated cardiomyopathy; ECG, electrocardiography; HCM, hypertrophic cardiomyopathy; LQTS, long QT syndrome; LVNC, left ventricular noncompaction; MRI, magnetic resonance imaging; SCD, sudden cardiac death; SQTS, short QT syndrome; VF, ventricular fibrillation.
In the case of nonsurvivors, clinical data were obtained on the circumstances of the death, cardiac arrest rhythm, events, and previous studies. Autopsies were usually performed by the Instituto de Medicina Legal (Institute of Legal Medicine) of Granada according to current guidelines and recommendations.19 Briefly, a macroscopic examination of the heart was performed to record weight, cavity size and thickness, coronary artery origin, trajectory, and lumen, valve anatomy, the presence of coronary thrombosis or information on acute myocardial ischemia, and a macroscopic and microscopic characterization of the myocardium. Biochemical, toxicological, and histological studies were performed in all cases. In addition, in cases with no definite diagnosis or, when deemed necessary by the investigator, in cases with a definite family member diagnosis, molecular autopsy was performed (Figure 1 and Annex 1 of the supplementary material) or genetic sequencing of a first-degree relative with evidence of the disease if no DNA was available from the deceased.
Cascade family screening was performed in the relatives of all cases with a confirmed clinical or genetic diagnosis.
Genetic TestingA genetic study was performed in most families, regardless of whether a clinical diagnosis was made. Some cases diagnosed with a cardiomyopathy or channelopathy were excluded if the genetic screening provided little clinical information and there was no apparent familial aggregation, such as in Brugada syndrome, left ventricular noncompaction, and some cases of dilated cardiomyopathy. DNA was obtained from peripheral blood samples stored with EDTA. The genetic study evaluated a series of genes (up to 242) linked to cardiomyopathy or channelopathy. These genes were sequenced using the Sanger method in studies performed before 2013; subsequently, the new NGS Illumina HiSeq 2500 system was used (see the technical details in Annex 2 of the supplementary material). Almost all cases found to be negative with Sanger sequencing were subsequently resequenced with NGS. All coding exonic and flanking intronic regions were sequenced. The pathogenicity of the variants detected was catalogued according to the previous description of each variant, in silico pathogenicity prediction using bioinformatics tools (Polyphen,20 SIFT,21 MutationTaster22), the degree of interspecies conservation of the residue, and the presence of the variant in public databases of the general population such as dbSNP and the NHLBI GO Exome Sequencing Project database. Finally, before a mutation could be considered probably pathogenic, it had to show coherence and familial cosegregation with the phenotype.
Statistical AnalysisA specific database was created for the study. Statistical analyses were performed using SPSS statistical software (version 20.0; Stata Corp, Chicago, United States). The Mann-Whitney U test was used for comparison of quantitative variables and the Fisher exact text was used for proportions. P < .05 was considered statistically significant.
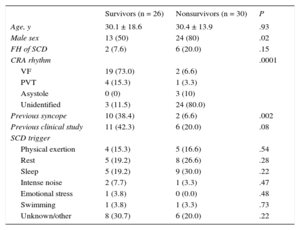
RESULTSThe final sample comprised 56 index cases or families (mean age of cases, 30.2 ± 16.1 years; 66.1% men). Of these, 53.6% were deceased probands (30 cases); the remainder were survivors (26 index cases). In the case of nonsurvivors, the cases referred to us were those suspected of having an inherited heart disease from a mean of 145 annual SCD episodes during the study period; of these retrospectively reviewed cases, up to about 15% were possibly due to an inherited heart disease. There was a higher prevalence of men in the nonsurvivor group, with no differences in age or other sociodemographic variables (Table 1). A cardiology study had previously been performed in 30.4% of the cases, most commonly due to syncope (21.4%).
Sociodemographic Characteristics of Sudden Cardiac Death Survivors and Nonsurvivors
| Survivors (n = 26) | Nonsurvivors (n = 30) | P | |
|---|---|---|---|
| Age, y | 30.1 ± 18.6 | 30.4 ± 13.9 | .93 |
| Male sex | 13 (50) | 24 (80) | .02 |
| FH of SCD | 2 (7.6) | 6 (20.0) | .15 |
| CRA rhythm | .0001 | ||
| VF | 19 (73.0) | 2 (6.6) | |
| PVT | 4 (15.3) | 1 (3.3) | |
| Asystole | 0 (0) | 3 (10) | |
| Unidentified | 3 (11.5) | 24 (80.0) | |
| Previous syncope | 10 (38.4) | 2 (6.6) | .002 |
| Previous clinical study | 11 (42.3) | 6 (20.0) | .08 |
| SCD trigger | |||
| Physical exertion | 4 (15.3) | 5 (16.6) | .54 |
| Rest | 5 (19.2) | 8 (26.6) | .28 |
| Sleep | 5 (19.2) | 9 (30.0) | .22 |
| Intense noise | 2 (7.7) | 1 (3.3) | .47 |
| Emotional stress | 1 (3.8) | 0 (0.0) | .48 |
| Swimming | 1 (3.8) | 1 (3.3) | .73 |
| Unknown/other | 8 (30.7) | 6 (20.0) | .22 |
CRA, cardiorespiratory arrest; FH, family history; PVT, polymorphic ventricular tachycardia; SCD, sudden cardiac death; VF, ventricular fibrillation.
Values represent No. (%) or mean ± standard deviation.
A final diagnosis was made in 80.4% of families, with no significant differences between the study groups (survivors vs nonsurvivors, 80.7% vs 80.0%; P = .53). Figure 1 shows how the definite diagnosis was reached in the 2 groups. ECG data were available for 100% of the surviving index cases vs 23.3% of the nonsurvivors. There were significant differences in the path to diagnosis in each family by study group: in nonsurvivors, the combination of the family study and the autopsy findings was crucial in most cases, whereas, in survivors, the diagnosis largely relied on the clinical tests performed in the proband. Nonetheless, data provided by the family study was vital for diagnosis in 1 case with idiopathic ventricular fibrillation in the proband and positive drug provocation testing in relatives. An autopsy was performed for 70% of cases and was considered conclusive in 76% of cases (either due to the identification of the underlying cardiomyopathy [11 cases] or due to being “blank” [5 cases]). In 5 cases, the autopsy obtained unclear, borderline, or incomplete results. The autopsy was performed by the Institute of Legal Medicine of Granada in 15 cases; in 6 cases, the autopsy was carried out in the Institute of Legal Medicine of the city in which the death occurred and we performed the family study.
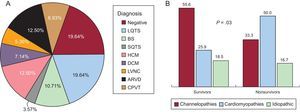
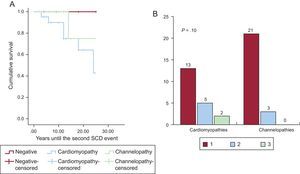
Figure 2 shows a schematic representation of the diseases found and their prevalence by study group. Channelopathy prevalence was significantly higher among survivors, largely due to long QT syndrome (8 cases). No definite diagnosis could be reached in a similar proportion of both groups. The diagnostic frequency of cardiomyopathy was significantly higher in nonsurvivors, largely due to hypertrophic and arrhythmogenic cardiomyopathy. In addition, the presence of a cardiomyopathy diagnosis was related, albeit nonsignificantly, to the probability of a second arrhythmic event in the family, studied from a retrospective point of view. Figure 3 shows a survival curve for the presence of a second arrhythmic event in a family, either SCD or ventricular arrhythmia or syncope. The probability more than 1 malignant arrhythmic event in an individual was 36.8% in families with a cardiomyopathy and 12.1% in families with a channelopathy (P = .10). Individuals diagnosed with channelopathy were significantly younger than those diagnosed with cardiomyopathy (26.0 ± 16.7 years vs 36.1 ± 16 years; P = .04).

A: Overall diagnoses obtained. B: A comparison between the 2 groups. ARVD, arrhythmogenic right ventricular dysplasia; BS, Brugada syndrome; CPVT, catecholaminergic polymorphic ventricular tachycardia; DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy; LQTS, long QT syndrome; LVNC, left ventricular noncompaction; SQTS, short QT syndrome.

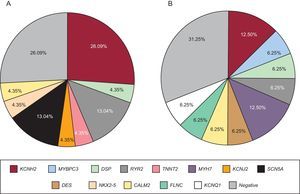
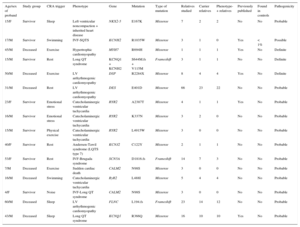
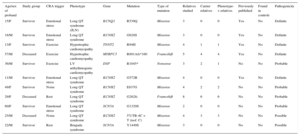
Genetic testing was performed in 71.4% of cases, either in the proband or in first-degree relatives with a positive phenotype if DNA from the deceased individual was unavailable. The type of genetic test performed depended on the phenotype and involved either NGS with specific selected panels of candidate genes (28 cases; 9 of these previously underwent Sanger sequencing) or exclusive use of the Sanger technique (12 cases). The genetic test was positive with identification of the probable causative mutation in 67.5% of cases but there were no significant differences according to the study group or sequencing method used. The most frequently mutated genes in both groups were KCNH2, RyR2, and DSP (Figure 4). Cascade screening was performed in 298 family members, a mean of 5.2 individuals per family, and identified 113 affected individuals. An example of cascade testing of relatives is shown in Figure 5. Table 2 and Table 3 detail cases from families who obtained positive genetic results using NGS and Sanger sequencing, respectively. In 9 of the cases analyzed using NGS, Sanger sequencing had previously obtained negative results. NGS allowed the diagnosis of 5 of these cases via identification of the causative mutation. Overall, genetic testing was key to the diagnosis of 12.2% of families and showed a trend for a better yield in individuals with channelopathies than in those with cardiomyopathies (95% vs 69% positivity; P = .08).
 and nonsurvivors (B).")
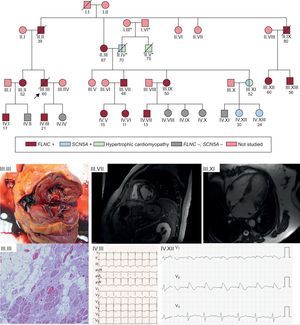
; this mutation was pathogenic and caused Brugada syndrome. The clinical figures in the lower part of the figure show the following: III.III: Cross-sectional macroscopic postmortem image of a left ventricular specimen showing ventricular dilatation and midwall fibrosis. In the microscopy image, abundant fibrosis and replacement of cardiomyocytes with adipose tissue. III.VII: A 48-year-old man with an FLNC mutation and extensive midmyocardial fibrosis of the left ventricle, findings that are similar to those seen in the autopsy of III.III. IV.III: ECG of a 21-year-old patient with an FLNC mutation and left ventricular cardiomyopathy. III.XI: hypertrophic cardiomyopathy in a 52-year-old woman without an FLNC mutation but with a Glu1225Lys mutation in SCN5A. IV.XII: Brugada type 1 pattern after flecainide in an asymptomatic daughter of the previous patient. ECG, electrocardiography; NGS, next-generation sequencing; SCD, sudden cardiac death. *, members of a different family from that which motivated the study; /, deceased subject; ○, woman; □, man; , proband.")
Example of a family with an FLNC p.Leu194fs mutation and a high rate of SCD events. Cascade screening identified 14 family members with the same mutation, with high levels of cosegregation for the arrhythmogenic cardiomyopathy phenotype. The cascade study identified the cause of a hypertrophic cardiomyopathy in a family member unaffected by the FLNC mutation but with another NGS-identified mutation in SCN5A inherited from the other branch of the family (his father); this mutation was pathogenic and caused Brugada syndrome. The clinical figures in the lower part of the figure show the following: III.III: Cross-sectional macroscopic postmortem image of a left ventricular specimen showing ventricular dilatation and midwall fibrosis. In the microscopy image, abundant fibrosis and replacement of cardiomyocytes with adipose tissue. III.VII: A 48-year-old man with an FLNC mutation and extensive midmyocardial fibrosis of the left ventricle, findings that are similar to those seen in the autopsy of III.III. IV.III: ECG of a 21-year-old patient with an FLNC mutation and left ventricular cardiomyopathy. III.XI: hypertrophic cardiomyopathy in a 52-year-old woman without an FLNC mutation but with a Glu1225Lys mutation in SCN5A. IV.XII: Brugada type 1 pattern after flecainide in an asymptomatic daughter of the previous patient. ECG, electrocardiography; NGS, next-generation sequencing; SCD, sudden cardiac death. *, members of a different family from that which motivated the study; /, deceased subject; ○, woman; □, man;
, proband.
Families With a Genetic Diagnosis Obtained Using Next-Generation Sequencing
| Age/sex of proband | Study group | CRA trigger | Phenotype | Gene | Mutation | Type of mutation | Relatives studied | Carrier relatives | Phenotype-+ relatives | Previously published | Found in controls | Pathogenicity |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 15/F | Survivor | Sleep | Left ventricular noncompaction + inherited heart disease | NKX2-5 | E167K | Missense | 7 | 2 | 2 | No | No | Probable |
| 17/M | Survivor | Swimming | IVF-SQTS | KCNH2 | R1035W | Missense | 3 | 1 | 0 | Yes | < 1% | Possible |
| 45/M | Deceased | Exercise | Hypertrophic cardiomyopathy | MYH7 | R694H | Missense | 1 | 1 | 1 | Yes | No | Definite |
| 15/M | Survivor | Rest | Long QT syndrome | KCNQ1 + KCNH2 | S644M.fs + V115M | Frameshift | 3 | 1 | 1 | No | No | Definite |
| 50/M | Deceased | Exercise | LV arrhythmogenic cardiomyopathy | DSP | R2284X | Missense | 8 | 4 | 4 | Yes | No | Definite |
| 31/M | Deceased | Rest | LV arrhythmogenic cardiomyopathy | DES | E401D | Missense | 66 | 23 | 22 | No | No | Probable |
| 23/F | Survivor | Emotional stress | Catecholaminergic ventricular tachycardia | RYR2 | A2387T | Missense | 4 | 1 | 1 | Yes | No | Probable |
| 16/M | Survivor | Emotional stress | Catecholaminergic ventricular tachycardia | RYR2 | K337N | Missense | 3 | 2 | 0 | No | No | Probable |
| 15/M | Survivor | Physical exercise | Catecholaminergic ventricular tachycardia | RYR2 | L4915W | Missense | 3 | 0 | 0 | No | No | Probable |
| 40/F | Survivor | Rest | Andersen-Tawil syndrome (LQTS type 7) | KCNJ2 | C122Y | Missense | 2 | 1 | 1 | No | No | Probable |
| 53/F | Survivor | Rest | IVF-Brugada syndrome | SCN5A | D1816.fs | Frameshift | 14 | 7 | 3 | No | No | Probable |
| 7/M | Deceased | Exercise | Sudden cardiac death | CALM2 | N98S | Missense | 3 | 0 | 0 | No | No | Probable |
| 16/M | Deceased | Swimming | Catecholaminergic ventricular tachycardia | RyR2 | L488I | Missense | 5 | 4 | 4 | No | No | Probable |
| 4/F | Survivor | Noise | IVF-Long QT syndrome | CALM2 | N98S | Missense | 3 | 0 | 0 | No | No | Probable |
| 60/M | Deceased | Sleep | LV arrhythmogenic cardiomyopathy | FLNC | L194.fs | Frameshift | 23 | 14 | 12 | No | No | Probable |
| 43/M | Deceased | Sleep | Long QT syndrome | KCNQ1 | R366Q | Missense | 16 | 10 | 10 | Yes | No | Probable |
CRA, cardiorespiratory arrest; F, female; IVF, idiopathic ventricular fibrillation; LV, left ventricular; M, male; SQTS, short QT syndrome.
Families With a Genetic Diagnosis Obtained using Sanger Sequencing
| Age/sex of proband | Study group | CRA trigger | Phenotype | Gene | Mutation | Type of mutation | Relatives studied | Carrier relatives | Phenotype-+ relatives | Previously published | Found in controls | Pathogenicity |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 15/F | Survivor | Emotional stress | Long QT syndrome (JLN) | KCNQ1 | R539Q | Missense | 3 | 0 | 0 | Yes | No | Definite |
| 18/M | Survivor | Emotional stress | Long QT syndrome | KCNH2 | G628S | Missense | 3 | 0 | 0 | Yes | No | Definite |
| 13/F | Survivor | Exercise | Hypertrophic cardiomyopathy | TNNT2 | R94H | Missense | 4 | 1 | 1 | Yes | No | Definite |
| 57/M | Deceased | Exercise | Hypertrophic cardiomyopathy | MYBPC3 | R891Afs*160 | Frameshift | 5 | 4 | 4 | Yes | No | Definite |
| 36/M | Survivor | Exercise | LV arrhythmogenic cardiomyopathy | DSP | R1045* | Nonsense | 3 | 2 | 1 | No | No | Probable |
| 11/M | Survivor | Emotional stress | Long QT syndrome | KCNH2 | G572R | Missense | 4 | 0 | 0 | Yes | No | Definite |
| 49/F | Survivor | Noise | Long QT syndrome | KCNH2 | E637G | Missense | 4 | 2 | 2 | No | No | Probable |
| 29/F | Deceased | Rest | Long QT syndrome | KCNH2 | G262fs | Frameshift | 8 | 6 | 6 | No | No | Probable |
| 60/F | Survivor | Emotional stress | Long QT syndrome | SCN5A | G1329S | Missense | 2 | 0 | 0 | No | No | Probable |
| 25/M | Deceased | Noise | Long QT syndrome | KCNH2 | 5′UTR-4C > T (isof. C) | Missense | 4 | 3 | 3 | No | No | Possible |
| 22/M | Survivor | Rest | Brugada syndrome | SCN5A | Y1449S | Missense | 3 | 0 | 0 | No | No | Possible |
F, female; JLN, Jervell and Lange-Nielsen syndrome; LV, left ventricular; M, male.
Our work shows that a systematic protocol can achieve a high diagnostic yield in SCD cases. There were no differences in the final diagnosis rates according to cardiac arrest survival or arrest trigger. However, there were differences in the path to diagnosis, with a high yield of autopsy and genetic studies in the relatives of nonsurvivors but a more heterogeneous path to diagnosis among survivors via the systematic application of conventional techniques in the proband. Genetic testing showed a high rate of positive results; although the difference was not significant, NGS was superior to Sanger sequencing.
Few studies have analyzed the differential characteristics of SCD cases according to survival from cardiac arrest. An Australian group11 found differences in the diagnostic yield between nonsurvivors and survivors, with a 3.5-fold higher rate of diagnoses in survivors. That study excluded patients with structural heart disease, in contrast to ours. The availability of a surviving proband is essential for the diagnosis of channelopathies due to the need for resting ECG, stress ECG, and ECG with epinephrine and flecainide. Our data agree with those conclusions because the prevalence of channelopathies was higher in survivors. A notable finding from our work was the high diagnostic yield of a combined autopsy and family study for cardiomyopathies, which enabled diagnosis in most cases. This finding differs from that of previous studies and might be due to various factors. On the one hand, we studied a considerable number of relatives per index case (5.2 relatives per index case), greater than that of previous works such as the Australian study (3.8 per case), the series of Behr et al.17 (3.2 per case), and the CASPER registry10 (1.3 per case). In addition, the retrospective nature of our series might have caused a selection bias due to the preferential study of cases with pathologic findings in the autopsy.
The higher prevalence of cardiomyopathies in the survivor group could indicate a worse prognosis of cardiac arrest in patients with cardiomyopathy vs those with primary electrical diseases. On the one hand, the mechanism precipitating the arrest could be different in channelopathies than in cardiomyopathies and, on the other hand, the normal myocardium of channelopathies might be less vulnerable to ischemia and more responsive to resuscitation maneuvers and defibrillation. This finding is supported by the greater proportion of patients with syncope in the survivor group, whereas the first event in nonsurvivors tended to be SCD. In addition, the rate of events in relatives was higher among patients with cardiomyopathies, with a higher prevalence of SCD and a trend for less time between these events. Similar findings have already been described in the follow-up of patients of the CASPER registry,10 which showed a higher frequency of appropriate therapies in the group of patients with cardiomyopathies. In addition, a more aggressive behavior has been proposed for primary electrical diseases.23
The differential characteristics of the 2 groups are also in line with those of previous work24 and our study contains the highest proportion of men in all series and, in particular, of nonsurvivors. There were no differences in the precipitating factors of the cardiac arrest, with a negligible incidence of intense exercise or competitive sport. This finding is important because individuals with cardiomyopathy are currently recommended to abstain from competitive physical activity, with slightly greater leeway for those with channelopathies, and thus the impact of exercise might not be as significant as once thought.25 In addition, because the precipitating factors of the cardiac arrest were similar, it is less likely that an individual's survival depends on the circumstances of the death.
A factor that significantly increased the diagnostic yield in both groups was the genetic study. Although this approach did help to reach a diagnosis in a relatively small number of index cases, its adequate use and interpretation enabled the identification of a considerable number of affected family members. Previous studies analyzed the usefulness of phenotype-directed genetic testing or testing of cases with no apparent phenotype.8–12 The classic approach to molecular autopsy is to sequence a small number of genes, such as KCNQ1, KCNH2, SCN5A, and RyR2,14–26 with expansion of the technique in specific cases to certain cardiomyopathies to enable cascade family screening. In the case of survivors of cardiac arrest without a phenotype, this approach is even discouraged by clinical practice guidelines.27 A previous study from our group indicated that application of the new technique of NGS to cardiac arrest survivors without a specific phenotype could increase the diagnostic yield.9 The present work provides additional evidence that genetic testing via NGS in both survivors and nonsurvivors (molecular autopsy) permits the identification of pathogenic mutations in less customary genes, such as CALM, FLNC, and DES, and allows cascade family screening. NGS represents a great opportunity in the SCD field due to its potential ability to diagnose cardiomyopathies and channelopathies28 and should be the genetic sequencing technique of choice for SCD.29 Given its ability to facilitate the analysis of multiple genes and improve sensitivity compared with Sanger, the challenge lies in the correct interpretation of the findings in order not to lose specificity, which is why an extensive cosegregation study is vital, as performed in our series. A recent prospective study with a large sample size determined a true usefulness of molecular autopsy using NGS of 27% for unexplained SCD.12
LimitationsAlthough a systematic study protocol was applied to cases referred to a specialized clinic, the retrospective design of the study suggests that some cases, particularly nonsurvivors, could have been missed due to the lack of a defined referral protocol and that certain cases could have been selected due to more evident suspicious findings of inherited heart disease in the autopsy. This would explain the difference between the nonsurvivors included and those that would be expected. This is also why 100% of the autopsy studies are not available: not all of the autopsies were performed by the Institute of Legal Medicine of Granada, which could lead to some interobserver variability. The retrospective collection of autopsy results meant that there were inconclusive findings in 5 cases and that the postmortem investigation could not be expanded. This limitation was minimized via rigorous single-center interpretation of the autopsy reports and the centralized and homogeneous study of all families in our unit. Another limitation is the sample size, which might be insufficient to reach more solid conclusions.
CONCLUSIONSA rigorous systematic protocol has a high diagnostic yield for SCD, with a higher prevalence of channelopathies among survivors and a worse prognosis in men and in families with cardiomyopathies. Genetic testing with NGS is useful in SCD cases, both those with and without an apparent phenotype, and its diagnostic yield is higher than that of the Sanger method.
CONFLICTS OF INTERESTNone declared.
- –
Nonischemic SCD is caused by inherited diseases and there may be concealed cases in relatives.
- –
Male sex is related to a higher prevalence of SCD. The underlying diagnosis has not been associated with the prognosis of relatives.
- –
Pharmacological testing, family screening, and molecular autopsy play a role in the study of unexplained SCD.
- –
The usefulness of genetic testing with conventional techniques in cases with an obvious phenotype has been shown but there is insufficient evidence regarding the value of NGS in the study of SCD.
- –
A definite diagnosis was reached via systematic study of probands and families in a high proportion of SCD cases.
- –
The incidence of cases among family members is high. A rigorous family study can be the key to diagnosis in both survivors and nonsurvivors of SCD.
- –
In families with a case of SCD, cardiomyopathy diagnosis appears to be associated with a higher event rate and worse prognosis.
- –
Genetic testing with NGS seems to be superior to the Sanger method due to its ability to detect mutations in less common genes.
First, we thank the participating families for their generosity at such a difficult time. We would also like to thank Dr C. Moro from the Institute of Legal Medicine of Seville for her valuable contribution to the histological study of the samples.