Keywords
INTRODUCTION
Long QT syndrome (LQTS) is characterized by severely altered ventricular repolarization, resulting in prolongation of the QT interval on electrocardiogram (ECG). The condition predisposes patients to malignant ventricular arrhythmia (torsade de pointes) and sudden death. The clinical and electrocardiographic description of long QT syndrome was reported in 1957 by Anton Jervell and Fred Lange Nielsen,1 who published their studies on a family of nonconsanguineous parents with 6 children. Four of the children had congenital deafness and syncopal episodes, and 3 presented sudden death. ECG study of these patients showed an unusually long QT interval. Both parents were asymptomatic, had a normal ECG, and presented no hearing problems. In 1964, Romano and Ward independently reported a cardiac syndrome characterized by recurrent syncope, a family history of sudden death, and prolongation of the QT interval without neuronal deafness.2 Later genetic studies showed that the syndrome described by Jervell and Lange Nielsen, which is accompanied by congenital neuronal deafness, corresponds to homozygous mutations, with a severe phenotype and high risk of sudden death. The condition known as Romano-Ward syndrome generally corresponds to heterozygous mutations, patients do not display hearing alterations, and the severity of the disease varies considerably. Almost half a century later, in 1995,3,4 the principal genes associated with LQTS were described and the disease was recognized as a cardiac ion channel disorder. It was the first cardiac channelopathy to be described and is perhaps the most extensively investigated arrhythmogenic ion channel disorder to date. The clinical picture varies greatly: the patient can be asymptomatic, or show recurrent syncope, seizures, or sudden death as the first manifestation of the disease. Initially, LQTS was considered a rare syndrome and, in effect, the severe presentation of the disease is sporadic. Nonetheless, the incidence of related mutations is estimated at 1/3000-5000 cases,5 32% of asymptomatic carriers can have a heart rate-corrected QT interval (QTc) within normal limits, the disease is transmitted to 50% of their descendants, they are more susceptible to develop arrhythmia when compared to the general population, and up to 20% can become symptomatic.6
Long QT syndrome displays great genetic heterogeneity. More than 500 mutations distributed in 10 genes have been described in this condition: KCNQ1, HERG, SCN5A, KCNE1, KCNE2, ANKB, KCNJ2, CACNA1, CAV3, and SCN4B. Despite the advances in this area, a genetic diagnosis cannot be established in 25%-30% of patients.7,8 The presentation of the disease is mainly monogenic6; polygenic or composite varieties usually have a more severe phenotype. Penetrance, ie, patients who have the mutation and manifest the phenotype, ranges from 25% to 90%.9 Less frequently, there may be variations in the expressivity of the disease, with several phenotypes resulting from the same mutation. Molecular genetic studies developed over the last 11 years have yielded important genotype-phenotype correlations, which have helped to guide the treatment approach. In addition, interesting observations have been made on individual susceptibility to developing arrhythmia in studies investigating the frequent nonsynonymous polymorphisms in this population, an aspect that has aroused considerable interest, particularly in the area of pharmacogenomics.
CLASSIFICATION OF LONG QT SYNDROME
General Concepts
The LQTS classification used in the past was based on the homozygous or heterozygous presentation of the disease, which gives rise to Jervell-Lange-Nielsen syndrome (with deafness) and Romano-Ward syndrome (without deafness), respectively. The present classification emphasizes the genetic findings, as is illustrated in Table 1. The 3 main genes associated with the disease were described in 1995-1996. These genes, which code for pore-forming units of the potassium channels IKs and IKr, and the sodium channel Nav1.5, account for nearly 65% of the cases. Although in subsequent years seven additional genes have been included in the list, they account for only 5% of the cases.

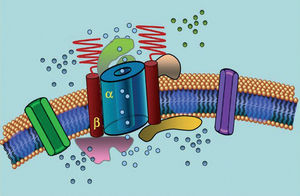
Ion channels are transmembrane proteins that transport ions through the cell membrane. The channels implicated in LQTS are selective or specialized in transporting a single ion and are voltage-dependent, ie, their activation occurs at a specific intracellular voltage, which varies according to the channel subtype. The electrical and contractile phenomena that occur in the cardiomyocyte are controlled by these structures. Ion channels form macromolecular complexes consisting of a main unit that forms the channel pore and auxiliary proteins that regulate it (Figure 1). The channel dysfunction seen in LQTS can occur at these two sites: the main protein or the regulating proteins (Table 1). Involvement of the pore-forming unit, known as alpha, generates the three most common subtypes of LQTS: LQTS1 (affecting the IKs potassium channel), LQTS2 (affecting the IKr potassium channel), and LQTS3 (affecting the sodium channel). Because these are the most frequent subtypes, they are the best characterized clinically and genetically. The phenotype-genotype correlations in these three main forms are described in Figure 2. Currently, Jervell-Lange-Nielsen syndrome corresponds to the LQTS 1 and 5 varieties. Characteristically, these patients have congenital deafness and compound homozygous or heterozygous mutations that affect the IKs current. Romano-Ward syndrome includes varieties from LQTS 1 to 10 and does not involve deafness.

Figure 1. Schematic representation of the macromolecular complex. The ion channels are transmembrane proteins (α) regulated by various proteins; one of them is the so-called β subunit.
Long QT Syndrome Type 1 (LQTS1)
Patients with LQTS1 usually present episodes of ventricular arrhythmia when exercising or when undergoing sympathetic stimulus (68%).10 Swimming has been described as a sport triggering arrhythmia in LQTS1.11 Penetrance is nearly 62% in this subtype. The T-wave in these patients often has a broad base and very prolonged duration12,13 (Figure 2). It is the most frequent subtype and explains 30%-35% of cases. The affected gene, KvLQT1 (or KCNQ1), is located on chromosome 11 (11p15.5) and codes for the IKs potassium channel α-subunit. The action potential is prolonged by a reduction in the outgoing K+ current during phase 3 of the action potential.

Figure 2. Genotype-phenotype correlation in the most frequent long QT syndromes. *Refers to cases that have the mutation and manifest the phenotype.
Long QT Syndrome Type 2 (LQTS2)
Patients with LQTS2 tend to present ventricular arrhythmia in response to emotional stress (49%) or sudden auditory stimuli (eg, an alarm-clock), and less frequently during sleep (22%) or exercise (29%).10 Women in the postpartum period are particularly susceptible.14 Estimated penetrance is 79%; hence, up to 20% of cases can have a nondiagnostic ECG. The T-wave in LQTS2 is usually low-amplitude and bifid, with notching12,13 (Figure 2). The affected gene is KCNH2 or HERG, located on chromosome 7 (7q35-36), which codes for the IKr potassium channel α-subunit and accounts for 25%-30% of cases. Dysfunction of this channel decreases the outgoing K+ current during phase 3 of the action potential, prolonging its duration.
Long QT Syndrome Type 3 (LQTS3)
Patients with LQTS3 have a greater risk of presenting malignant arrhythmias during rest (sleep) or bradycardia.15 Penetrance of the SCN5A gene mutation is nearly 90%. The ECG in LQTS3 usually shows a delayed, pointed T wave and allows clear observation of the ST segment prolongation12,13 (Figure 2). These patients usually have fewer symptoms than those with LQTS1 or LQTS2, but the events are characteristically more lethal.
The affected gene in LQTS3 is SCN5A, which codes for the Nav1.5 sodium channel α-subunit (Figure 1), located on chromosome 3 (3p21-24); it is the cause of disease in 5%-10% of the cases. Defective inactivation of the channel allows sustained input of Na+ during phase 2 of the action potential, prolonging its duration.
Long QT Syndrome Type 4 (LQTS4)
Type 4 is a rare variety of LQTS, accounting for nearly 1% of cases. It is an atypical form that produces a wide spectrum of arrhythmias, including catecholaminergic polymorphic ventricular tachycardia, atrial fibrillation, intraventricular conduction alterations, sinus node dysfunction, and bradycardia6-18; in addition, the QTc can be within normal limits in many patients. The affected gene is ANKB, located on chromosome 4 (4q25-27), which codes for synthesis of ankyrin-β, a structural protein that links cardiomyocyte membrane proteins to cytoskeletal proteins. These proteins are the Na/K ATPase pump, Na/Ca exchanger, and inositol triphosphate receptor (InsP3R). Mutations causing a loss of ankyrin-β function lead to increases in intracellular calcium concentration and alterations in the expression of N/K ATPase and Na/Ca exchanger. The elevated calcium concentration gives rise to early and delayed after-depolarizations. Thus, the ventricular arrhythmias observed in ankyrin-β gene mutations are due to spontaneous depolarizations, usually in response to catecholaminergic stimulation.
Long QT Syndrome Type 5 (LQTS5)
Type 5 originates with changes in the sequence of the KCNE1 gene located on chromosome 21 (21q22.1p22.)19 KCNE1 codes for synthesis of the IKs channel β-subunit, also known as the minK subunit, which regulates the IKs channel. This type accounts for less than 1% of cases.
Long QT Syndrome Type 6 (LQTS6)
The affected gene in type 6 is KCNE2, located on chromosome 21 (21q22.1).20 This gene codes for the potassium channel β-subunit, also known as the MiRP1 subunit, and it regulates the IKr channel. Less than 1% of cases are type 6.
Long QT Syndrome Type 7 or Andersen-Tawil Syndrome (LQTS7)
The dysmorphic findings and electrocardiographic alterations seen in this syndrome were first described in 1971 by Dr Andersen21 and revisited in 1994 by Dr Tawil,22 but the genetic/molecular description was not reported until 2001.23 Now known as Andersen-Tawil syndrome (ATS) this condition is an autosomal dominant alteration characterized by periodic paralysis, abnormal skeletal development, ventricular arrhythmia of the type involving frequent ventricular extrasystoles, and a particular susceptibility to develop ventricular fibrillation, particularly in women. The alterations described in ATS include ventricular extrasystoles (41%), nonsustained polymorphic ventricular tachycardia (23%), bidirectional ventricular tachycardia (68%), and torsade de pointes (3%).24 Some of the observed dysmorphic characteristics include short stature, scoliosis, clinodactyly, hypertelorism, low implantation of the ears, micrognathia, and a wide forehead. Disease expression varies, a fact that complicates early diagnosis.23,25 Mutations in the KCNJ2 gene located in chromosome 17 (17q23), which codes for synthesis of the rectifying potassium channel Kir 2.1, accounts for 70% of cases. This channel participates in phase 4 of the action potential. Several authors question the inclusion of this gene within the LQTS causal group, because the QTc interval is only slightly prolonged in this syndrome or even normal, but the U wave is usually prominent, which has led to overestimation of the QT interval. The reader will find that some authors suggest that KCNJ2 mutations generate ATS1 and not LQTS7.24
Long QT Syndrome Type 8 (LQTS8)
Type 8 arises from mutations in the CACNA1 gene located on chromosome 12 (12p13.3), which codes for L-type calcium channel Cav1.2. It causes Timothy syndrome,26 a condition characterized by cardiac malformations, intermittent immunological deficiency, hypoglycemia, cognitive alterations including autism, interdigital fusion, and prolonged QT, which leads to cardiac arrhythmia and sudden death.27 Less than 0.5% of cases are type 8.
Long QT Syndrome Type 9 (LQTS9)
This variety of LQTS develops from mutations in the CAV3 gene, located on chromosome 3 (3p25), which codes for caveolin 3 synthesis. The caveola is an invagination of the plasma membrane implicated in endocytosis, lipid homeostasis, and signal transduction. An important component of this structure is caveolin, which has 3 known subtypes; subtype 3 is specific for skeletal and cardiac muscle. Some ion channels are co-located in the caveola, including a cardiac isoform of sodium channel Nav1.5. Several mutations in this protein have been recently described. These alter the biophysical properties of sodium channel Nav1.5 in vitro, generating a phenotype similar to that observed in LQTS3.28 Less than 1% of cases are attributed to this cause.
Long QT Syndrome Type 10 (LQTS10)
Type 10 was described in a very severe case, with QTc >600 ms, fetal bradycardia, and 2:1 atrioventricular (AV) block. It results from mutations in the SCN4B gene, located on chromosome 11 (11q23), which codes for the sodium channel β4-subunit. Four different subtypes of β subunits have been described, which interact and regulate the various sodium channel isoforms; nonetheless, only subtype 4 has been associated with arrhythmogenesis up to now.29 The incidence of mutations of this gene has not been examined, but is estimated at <1%.
Mutations of the Jervell-Lange-Nielsen Variety
This severe form of LQTS is caused by homozygous30 or compound heterozygous mutations of the KCNQ1, and/or KCNE1 genes, which code for the IKs current; ie, a very severe variety of the LQTS1 or LQTS5 forms. This condition is characteristically associated with congenital deafness. Patients usually have a QTc>500 ms and recurrent syncope, and are at a high risk for sudden death. The parents of patients with this variety are usually heterozygous and have less severe disease, or show no symptoms.31
DIAGNOSIS OF LONG QT SYNDROME
Schwartz Score
In 1985, Schwartz et al32 published the criteria for diagnosing LQTS, which were modified in 1993 and contain important guidelines for the initial evaluation of potential cases. This system uses a score of 1 to 9 based on the family history, and the clinical and electrocardiographic findings. The probability of disease is low at a score of ≥1, intermediate at 2-3, and high at ≥4 (Table 2).

Prenatal Diagnosis of Long QT Syndrome
Fetal bradycardia can be one of the first clinical manifestations of LQTS. Retrospective series have shown that up to 70% of patients diagnosed with LQTS during childhood have a history of bradycardia, usually accompanied of fetal hydrops.33 Assessment of fetal cardiac repolarization between weeks 14 and 39 is useful for early diagnosis of LQTS.34
Gonadal mosaicism for LQTS has been associated with recurrent fetal losses during the third trimester of pregnancy.35 If the disease is highly suspected, amniocentesis after 16 weeks of gestation can be useful for establishing the diagnosis, which is easily reached when one of the parents is known to be the carrier of a specific mutation.36
STUDY OF A PATIENT WITH LONG QT SYNDROME
Clinical History
A family and/or personal history of sudden death is of crucial importance for both the diagnosis and risk stratification of LQTS. In addition, precipitating factors and the context of syncope can indicate the LQTS subtype. In the initial evaluation of a suspected case, the use of drugs that can prolong the QT interval should be ruled out.
QT Interval: What Is Normal?
The QT interval should be measured preferentially in leads II or V5,37 where it has been proven to have greater predictive value.38 This interval indicates the duration of ventricular repolarization and is measured from the beginning of the Q wave to the end of the T wave. Conventionally, the formula proposed by Bazett39 is employed to correct the duration of the interval according to the heart rate (QTc=QT/√RR, expressed in seconds). Although measurement of the QT interval seems simple, in a multicenter study carried out by Viskin et al,40 less than 40% of physicians other than cardiologists, less than 50% of cardiologists, and more than 80% of specialists in arrhythmia knew how to measure it properly. It is advisable for physicians to carry out manual measurement and not trust automated measurements, which may be useful for other intervals, but are imprecise when calculating the QT interval. The QT is a dynamic interval and the normal limits depend on several factors. Although a QTc interval of é440 ms in males and é460 ms in females is considered abnormal, one can find carriers of mutations as well as healthy individuals within this range (Figure 3). In families with LQTS1, Vincent et al41 demonstrated that none of the cases with a positive genotype had a QTc<410 ms and none with a negative genotype had a QTc>470 ms. Monnig et al38 recently showed that QTc>440 ms suffices to detect patients with LQTS-associated mutations, QTc>470 ms is useful to identify patients at risk of developing symptoms, and QTc>500 ms is found in symptomatic patients undergoing treatment.

Figure 3. Model showing distribution of the heart rate-corrected QT interval (QTc) in patients with mutations in KVLQT1, HERG, or SCN5A, and their unaffected family members. The curve to the left describes distribution of unaffected members and the curve to the right, affected members.
Other Electrocardiographic Alterations Associated With Long QT Syndrome
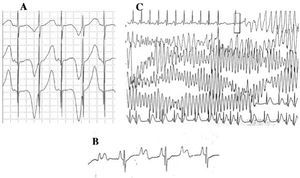
Patients with LQTS can present multiple T wave alterations: polarity alternans, amplitude variations, notching, and a biphasic appearance, among others.42 T wave alternans (Figure 4A) is defined as a beat-by-beat variation in amplitude, morphology and polarity of a sinus rhythm T wave, without variations in the QRS complex. It is an indicator of electrical instability,43 reflecting regional dispersion of ventricular repolarization, and occasionally precedes ventricular fibrillation.44

Figure 4. Electrocardiographic alterations in long QT syndrome. A: T wave electrical alternans. B: atrioventricular block 2:1. C: self-limited torsade de pointes.
Patients with LQTS can progress with signs of sinus node dysfunction, bradycardia, and/or pauses.45 The LQTS1 and LQTS3 subtypes, particularly the latter, often present sinus bradycardia,46 whereas LQTS4 has been associated with sinus node dysfunction.18
Since the decade of 1970-1980, the coexistence of AV conduction defects with LQTS47 has been observed (Figure 4B). Two-to-one AV block is an infrequent manifestation with a poor prognosis that can be present since the fetal stage in the form of persistent bradycardia. The incidence of this abnormality has been reported at 4%-5%48 and it is associated with high mortality despite treatment with beta-blockers and/or pacemakers.49,50 This phenomenon can be explained by a lengthy duration of the action potential. When the ventricular refractory period is extended, the following impulse of sinus activity is blocked because it reaches the ventricles when they are still in the refractory period. This alteration seems to occur exclusively in LQTS, because the ventricular refractory period is greater than that of the AV conduction system.51 The slope of the QRS complex is usually steep and the block has been localized in the infraHis area,46,51,52 but the site of the block may depend on the genotype. Up to now, 4 genes have been related to 2:1 block in LQTS: HERG (LQTS2),53,54 SCN5A (LQTS3),52 CACNA1 (LQTS8),26 and SCN4B (LQTS10).55
The characteristic ventricular arrhythmia of LQTS is known as torsade de pointes (Figure 4C). It presents when the QT interval is prolonged, regardless of the etiology. It is a polymorphic ventricular tachycardia due to reentry, characterized electrocardiographically by continuous twisting of the QRS axis around an imaginary line. It is commonly preceded by a pause followed by an extrasystole (short-long-short RR interval), as is shown in the figure.56-58 It can culminate in ventricular fibrillation and sudden death. If this does not occur, the patient may only experience syncope, and if the episode is brief, it may go undetected.
Holter
Holter study provides a complete, dynamic assessment of the QT interval. Occasionally spontaneous episodes of asymptomatic ventricular arrhythmia are recorded, as well as episodes of sinus node dysfunction or AV block.
Exercise Stress Test
Patients with LQTS cannot reach the maximum expected heart rate calculated according to age. In addition, under exertion the QT interval can display paradoxical behavior, by increasing rather than decreasing.59,60 The electrocardiographic pattern during exercise stress testing will be different depending on the type of LQTS. Patients with LQTS1, in addition to not reaching the maximum calculated heart rate for their age, frequently show an increased QT interval, while those with LQTS2 can reach their expected heart rate and show only a mild QT interval increase or none at all.61,62 In general, patients with LQTS3 have a physiologic response to exercise, ie, normal shortening of the QT interval.63 Stress testing can also be useful for assessing treatment response and for stratifying risk in asymptomatic cases, or when there are doubts as to the events leading to arrhythmia.
Genetic Screening
In the last years, genetic studies in LQTS have been limited to research laboratories. Nevertheless, the information derived from these efforts has been extremely useful for treating patients, particularly high-risk cases. Perhaps the main application of screening is in genetic counseling, but it also has important implications in treatment, which can be oriented according to the affected channel. The precise location of a given mutation can provide additional information regarding the evolution of risk. Patients with mutations in the transmembrane region of KCNQ1 (IKS) have a greater probability of presenting arrhythmic events than those with mutations in the C-terminal region64; the same is true for patients with mutations in the pore region of KCNH2 or HERG65 as compared to those with mutations in the N- or C-terminal regions.66
Initial screening may perhaps be limited to the KCNQ1, HERG, and SCN5A genes, which provide the possibility of encountering mutations in 65% of cases. When the results obtained are negative, screening can be extended to the KCNE1, KCNE2, ANKB, KCNJ2, CACNA1, CAV3, and SCN4B genes, which will increase the possibility of positive results by 5% to 10%.
Postmortem Genetic Screening
It is interesting that gene mutations leading to LQTS have been found in children who experienced sudden death and in inexplicable cases of sudden death in young adults.
Postmortem genetic studies of patients with sudden death and negative autopsy have shown mutations leading to LQTS in varying percentages67-69: close to 10% in children and 35% in young adults.70-72 Based on these results, routine ECG study has been proposed in all newborns.73,74
Postmortem genetic study, also known in the literature as "molecular autopsy," in addition to legal repercussions, has important implications in families who might be affected without their knowing it.
Regulatory Polymorphisms
Several frequently occurring polymorphisms have been described in the LQTS population, distributed in nearly all the genes associated with this condition. Although these changes are apparently not pathogenic, some can have the following effects75-78:
1. Generate individual susceptibility to develop arrhythmia.
2. Favor the pathogenic impact of another nonsynonymous change.
3. Decrease the pathogenic effect of another nonsynonymous change.
This is the case of the K897T polymorphism in KCNH2 (HERG), which is present in up to 15% of the population and is not only linked with susceptibility to certain drugs,79 but also favors the pathogenic effect of mutations in the same gene.78 Another example is the S1103Y polymorphism in the SCN5A gene, found mainly in blacks, which has an incidence of nearly 13% and is associated with an increased risk of sudden death in childhood.80
Interestingly, two alternative processing sites generating two types of sodium channels have been described in the product of the SCN5A gene, (which codes for the Nav1.5 sodium channel isoform in humans): one with 2016 aminoacids containing glutamine in the 1077 (Q1077) position, and another with 2015 aminoacids lacking glutamine (Q1077del). Transcripts of these alternative processings are present in a 2:1 proportion in the same human heart and several frequent polymorphisms will have different effects on channel functioning, depending on whether the context is Q1077 or Q1077del. This was initially shown with the H558R polymorphism of SCN5A, present in up to 30% of the population. When H558R was expressed in the context of Q1077, a profound reduction in the ion current was observed.81 A similar effect was documented with the S524Y82 polymorphism. These findings have provided factors to explain the varying severity of the disease, as well as the different phenotypes of the same mutation observed in some families.77
Pharmacological Testing With Adrenaline
Pharmacological testing with low-dose adrenalin is a safe, useful option to unmask suspected cases of LQTS with a borderline QTc. It is particularly effective for detecting asymptomatic forms of LQTS1, with a sensitivity of 92.5%, specificity of 86%, positive predictive value of 76%, and negative predictive value of 96%. It can also be useful in the diagnosis of LQTS2, with lower sensitivity and specificity. It is not useful for LQTS3 or other forms of LQTS. Under normal conditions, sympathetic stimulation induces phosphorylation of the IKs potassium channel, optimizing its function and giving rise to shortening of the action potential. In patients with LQTS, in particular type 1, a paradoxical response to administration of low-dose adrenalin (0.025-0.2 µg/kg/min) that prolongs the QT interval to more than 30 ms83-86 is observed.
QT INTERVAL PROLONGATION AND DRUG-INDUCED TORSADE DE POINTES
A great variety of drugs used in different medical specialties can cause a iatrogenic increase in the QT interval. Some drugs have been removed from the market because of this undesirable effect (eg, astemizole and cisapride, among others; for more information, visit www.qtdrugs.org).87,88
Ventricular arrhythmia secondary to non-antiarrhythmic drugs occurs in less than one of every 10 000 to 100 000 exposed subjects. Considering that clinical studies include between 2000 and 3000 subjects, this undesirable and fatal adverse event would easily escape detection during the clinical phase of drug development.89 This point has generated enormous interest in aspects referring to safety in the study and development of new drugs.
The factors related to individual susceptibility include female gender, hypocalcemia, hypomagnesemia, bradycardia, heart failure, postcardioversion, atrial fibrillation, left ventricular hypertrophy, undetected LQTS, predisposing polymorphisms, and high serum concentrations of predisposing drugs.90
The channel that typically interacts with drugs is IKr, coded by the KCNH2(HERG) gene, because of its molecular structure. Other potassium channels have 2 proline residues angled toward the channel pore, reducing its lumen. In contrast, IKr lacks these residues, a larger pore vestibule is generated, and exposure to large molecules is facilitated. In addition, it has 2 aromatic residues (tyrosine and phenylalanine) that favor binding with aromatic molecules present in several drugs able to block the channel.91
As was mentioned above, LQTS penetrance is incomplete and some asymptomatic carriers of mutations might manifest malignant arrhythmia upon receiving one of these drugs. In addition, polymorphisms considered frequent in the population confer individual susceptibility to the development of torsade de pointes when some drugs are used. This is the case of the R1047L polymorphism, the second most frequent in KCNH2, which has been associated with the development of torsade de pointes with use of the drug dofetilide.92 At least 20 KCNH2 gene polymorphisms have been described in healthy persons and their effect in individual susceptibility to develop drug-related arrhythmia remains to be determined.93 Polymorphisms that confer susceptibility to the development of ventricular arrhythmia have also been documented in sodium channel Na1.5. This is the case of the H558R polymorphism, which is present in up to 30% of the population, or S1103Y, which is frequent in blacks80,81,90,94,95; their implication in drug-induced susceptibility has not been investigated.
LONG QT SYNDROME AND PREGNANCY
Genetic counseling is important in LQTS, but in general terms there is no contraindication for pregnancy in women who are carriers, although each case is different and should be assessed individually in the appropriate context.
It has been noted that the risk of presenting malignant ventricular arrhythmia decreases with pregnancy. In contrast, greater vulnerability to present malignant arrhythmia has been reported within the first 9 months after delivery, particularly in patients with LQTS2. This risk decreases considerably with beta-blocker therapy.96
RISK STRATIFICATION
The evolution of LQTS varies and is influenced by the duration of the QTc interval, environmental factors, age, genotype, and response to treatment.97,98 Ventricular arrhythmia is more frequent in LQTS1 and LQTS2, but is more severe in LQTS3.99 As was mentioned above, women are especially susceptible to malignant arrhythmia during the postpartum period.14
Long QT syndrome should be considered high-risk when it is associated with the following:
1. Congenital deafness (Jervell-Lange-Nielsen syndrome).
2. Recurrent syncope due to malignant ventricular tachyarrhythmia.
3. Family history of sudden death.
4. QTc>500 ms.
5. 2:1 atrioventricular block.
6. T wave electric alternans.
7. LQTS3 genotype.
The study by Priori et al97 performed in 647 patients showed that the probability of presenting a major event (syncope, cardiac arrest, sudden death) before 40 years of age is high (>50%) when QTc is >500 ms in LQTS1, LQTS2, and in males with LQTS3. Recently, an analysis of the international LQTS registry was reported. The risk of sudden death was analyzed in 2772 adolescents with the disease, and 3 factors associated with higher risk in this population were identified: QTc>530 ms, history of syncope in the past 10 years, and gender; 10 to 12-year-old boys had a higher risk than girls, but in the 13 to 20 age range, the risk was comparable.100
TREATMENT
Symptomatic patients who do not receive treatment have a yearly mortality rate of 20% and 10-year mortality of 50% after a first event of ventricular arrhythmia. Although it is clear that treatment should be established when there are symptoms, the approach to use in asymptomatic patients is still under debate. It has been documented that cardiac arrest may be the first manifestation of the disease in 9% of patients,48 and that 12% of asymptomatic patients will develop symptoms and may experience sudden death. Initial treatment with beta-blockers should be started in all patients with LQTS. Exercise restriction is recommendable, but the clinical and electrocardiographic risk markers are a useful basis for decision-making. It is important to inform patients about the risk of using several drugs that can prolong the QT interval and favor the development of ventricular arrhythmia, as is mentioned above. Genetic diagnosis, apart from allowing appropriate family counseling related to the disease, is a help for assessing the prognosis and orienting specific treatment.
Beta-Blockers
Beta-blockers are the first-line treatment for LQTS and all patients should receive them as the initial therapy.101 They provide a reduction in the risk of cardiovascular events of up to 64%100 and are particularly effective in patients with IKs channel mutations (LQTS1),102 which are regulated to a great extent by the sympathetic system. Beta-blockers do not modify the QT interval, but instead, its dispersion.103 Although these drugs decrease the incidence of events,104,105 it has been shown that 10% of patients with LQTS1, 23% with LQTS2, and 32% with LQTS3 will have cardiovascular symptoms despite treatment.106 Patients with LQTS3, in particular, do not seem to obtain important benefits; in fact, this drug group should be used with caution in these patients, because episodes of ventricular arrhythmia in LQTS3 are more common when the heart rate is low. In general terms, 32% of symptomatic patients will have recurrent symptoms in the first 5 years before beginning beta- blocker treatment, and 14% of patients rescued from a sudden death episode will present another similar event within 5 years if they receive only this therapy.107 Several beta-blockers have been used in the treatment of LQTS, mainly nadolol (0.5-1 mg/kg/day), propranolol (2-4 mg/kg/day), metoprolol (0.5-1 mg/kg/day), and atenolol (0.5-1 mg/kg/day). Atenolol may not be beneficial in LQTS, however; it has been notified that at least 75% of patients who did not respond to beta-blocker therapy were receiving atenolol, although this finding may be related to the use of suboptimal doses.104 Exercise testing is useful to establish the appropriate dose. Maximum heart rate should not exceed 130 beats/min during treatment.
Sodium Channel Blockers
Sodium channel mutations that cause LQTS3 produce defective inactivation of the channel; sodium channel block has proven to be useful in these patients. Studies done with flecainide have documented improvements in the heart rate, T wave alterations, and QT interval.108 Mexiletine has also been reported to improve the electrocardiographic risk markers.63,109,110 In vitro studies with ranolazine have shown decreases in the deleterious effects of mutations reported in humans.111 Although the results are encouraging, it should be kept in mind that there are no long-term studies assessing this therapy, and no reported findings from large series. Sodium channels blockers should not be administered if there is no confirmed genetic diagnosis.
Potassium Supplementation and Drugs That Increase Its Availability
Potassium supplements and/or potassium-sparing drugs, such as spironolactone, shorten the QTc interval in 24% of cases.112,113 Drugs that favor opening of the potassium channels, such as aprikalim, levcromakalim, nicorandil, and pinacidil, have shown to be useful in the treatment of LQTS. The subtypes in which they are of particular benefit are LQTS1 and LQTS2.114
Pacemakers and Defibrillators
Pacemaker stimulation has been used in patients with pause-dependent arrhythmia.115,116 Patients with LQTS3 usually benefit more from this treatment because the prevalence of bradycardia is greater in this group. DDD pacing is indicated in patients with pause-dependent arrhythmia or high-grade 2:1 AV block. Frequencies programmed below 70 beats/min117 are not useful for preventing ventricular arrhythmia. It is recommended to program the sensor to fast response, because these patients usually have inappropriate heart rate acceleration in response to exercise. All functions that imply the presence of pauses should be shut off, such as the hysteresis and nocturnal function. The PARP (postventricular atrial refractory period) should be as short as possible. The frequency regulation function should be on to prevent postextrasystolic pause. It should be remembered that T wave oversensing and capture failures can also give rise to pauses. Combined use of an implantable cardioverter defibrillator (ICD) and beta-blockers substantially decreases the incidence of sudden death.118-120 The indication for these measures is clear in high-risk cases.121 Programming of the device will vary according to the needs of the individual patient, but, generally, administration of treatment in asymptomatic, self-limited events should be avoided; to this end, a detection time of 15 s is indicated. Arrhythmic storm is a complication of AID therapy. Nearly 15% of patients can experience this complication, which is due, in good part, to increased sympathetic tone following the ICD shock.118 This problem can be managed by increasing the beta-blocker dose. If this measure is not useful, resection of the sympathetic chain ganglia should be considered.
Left Sympathectomy
In 1971 sympathetic gangliectomy was introduced as a useful therapeutic option in these patients.122 In 1991, Schwartz et al123 published the first series of 85 patients with a poor response to beta-blocker treatment, in whom a left stellectomy was performed with encouraging results: a 5-year survival rate of 94%. Currently, this therapeutic option is offered to high-risk patients who persist with syncope despite beta-blocker treatment and/or pacemaker implantation, and those who experience frequent shocks from their implanted defibrillator. The procedure consists of resection of the inferior portion of the stellate ganglion and the T2 to T4 left thoracic ganglia of the sympathetic chain, since simple left stellectomy has not proven sufficiently effective. Microinvasive thoracoscopy124,125 has been used with good results. The largest series of patients treated with this method was recently reported and showed a significant reduction in the number of syncope episodes or sudden deaths, as well as a 5-year survival rate of 95%. In patients with previous syncope, 5-year survival was 97%, with an 11% possibility of recurrence, which, in the majority, consisted of a single syncopal event. There was also a significant reduction in the QT segment following left sympathectomy. Despite these favorable results, prevention of sudden death is not complete, but has been reduced to 3%. In patients with an ICD who underwent surgery because of multiple defibrillator shocks, the mean number of events decreased from 25 to 0, a 95% reduction. A beneficial effect was confirmed in LQTS1. Benefits are likely to be smaller in patients with LQTS2, and in LQTS3, its effectiveness has not been proven.126
Ablation
It has been reported that ablation of the extrasystole, which in some cases initiates the ventricular arrhythmia, can be carried out with a reduction in the incidence of episodes.127 However, there are no long-term studies with an appropriate number of patients to justify routine use of this technique.
See editorial on pages 675-82
ABBREVIATIONS
AV: atrioventricular
AID: automatic implantable defibrillator
ECG: electrocardiogram
QTc: heart rate-corrected QT
ATS: Andersen-Tawil syndrome
LQTS: long QT syndrome
Dr. Medeiros receives economic support from CONACyT and FUNSALUD.
Correspondence:
Dra. A. Medeiros-Domingo.
Unidad de Biología Molecular. Instituto Nacional de Ciencias Médicas
y Nutrición Salvador Zubirán.
Vasco de Quiroga, 15.
Tlalpan 14000. Mexico DF. Mexico.
E-mail: argeliamed@yahoo.com